Thank you for uploading that image.
By the way, files that are less than 50MB can be attached to this support request, which you might find more convenient than uploading.
If you change settings that affect the extraction width, the only way to see the results of that change would be by doing a "Reimport".
"Rescore" can be used for things that happen after chromatogram extraction, but which affect peak picking, such as changing the "Internal standard type" at Settings > Peptide Settings > Modifications.
When you do a reimport, or a rescore, any peak boundaries that you have manually adjusted will remain the same. For peaks that have not been user-modified, the boundaries might change if the new chromatogram data causes Skyline to pick a different peak.
For SureQuant methods, we recommend that you go to:
Settings > Transition Settings > Instrument
and check the checkbox that says "Triggered chromatogram extraction".
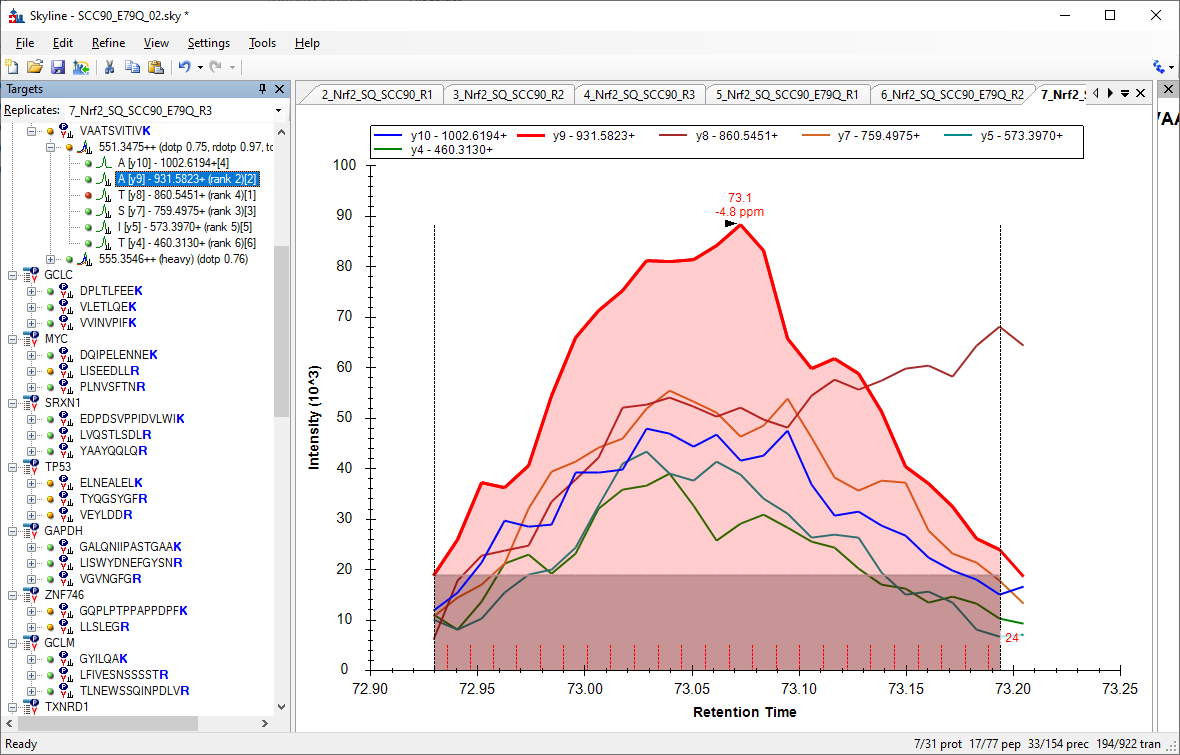
The major reason that you should do that is that Skyline will incorrectly estimate the background if the start and ends of the peak are truncated.
You can learn more about the triggered acquisition setting here:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=TriggeredAcquisition
Attached is a screenshot showing that the background for the y9 chromatogram is very large (shaded rectangle at the bottom of the peak).
Here is a page that talks about how Skyline usually calculates background (when "triggered acquistion" is off):
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tip_peak_calc
Also, if you click on points along a chromatogram, Skyline will bring up the full scan graph, which shows you a spectrum, and also shows the m/z channel that Skyline is summing across when extracting the chromatogram. In the attached image "Full Scan Graph", it looks like the y8 transition has interference from something that will not be able to be filtered out no matter how narrow you make the extraction width. When you have interference like this, it is recommended that you mark the transition as "Non quantitative", and then its area will not be included in things like "Total Area". You can mark a Transition as non-quantitative by right-clicking on it in the Targets tree. (or, you might want to simply delete that transition from your Skyline document)
One more thing: I noticed that at "Settings > Transition Settings > Filter" you have "Product Ion Selection > To" set to "last ion - 1". We normally recommend that you set that to "last ion". In a peptide with 8 amino acids in it, the "last ion" is y7 or b7. Some people choose "last ion - 1" because they incorrectly think the last ion would be y8 or b8, but that's not how Skyline counts things. There are a lot of example Skyline documents out there that are still using "last ion - 1" because we haven't been very good at spreading the word about what the recommended setting is.
-- Nick
 y9background.png
y9background.png FullScanGraph.png
FullScanGraph.png