| Brendan MacLean responded: |
2021-07-12 07:19 |
Surprising. What kind of spectra? Centroided or profile mode? Orbi, TOF, or QIT?
What are your Transition - Full-Scan settings?
|
| |
| roman sakson responded: |
2021-07-12 07:37 |
Hi Brendan,
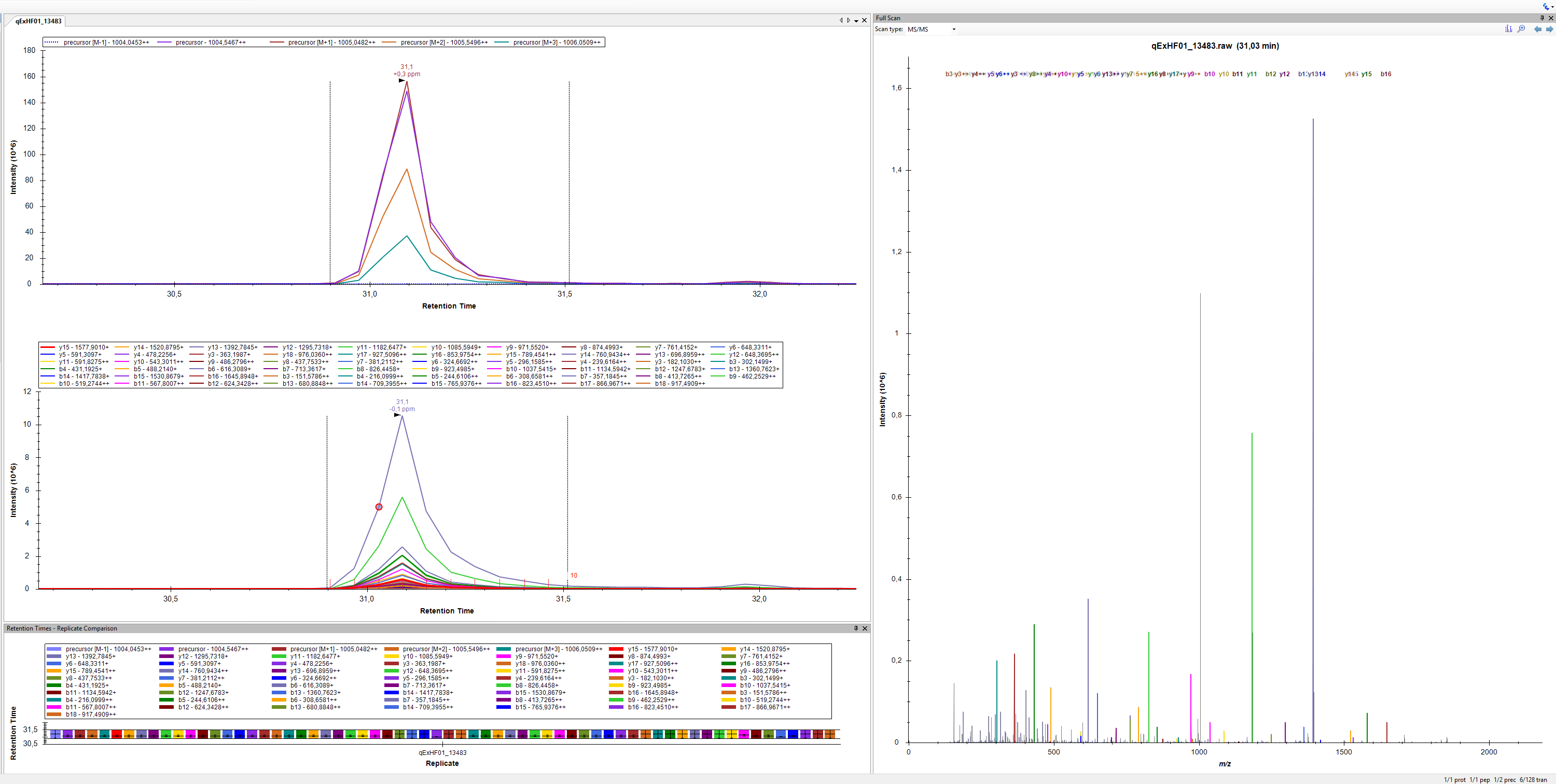
it is MS1+PRM on an Orbitrap QE HF (see attached). I have combined several measurements which were acquired with slightly different resolution settings within the same document (I adjusted the settings accordingly before importing the respective file). Not quite sure, whether Skyline likes that but XICs always looked fine to me. I still like to look at profile data during method development, especially for 120 k resolution... Would you recommend to go to centroided mode instead?
|
|
| |
| Brendan MacLean responded: |
2021-07-12 09:22 |
I wouldn't use library ion match tolerance is 0,5 m/z (500 ppm at 1000 m/z) with this case. I would recommend 0,05 m/z or lower (50 ppm at 1000 m/z).
Though, I am not totally sure what is going on with this document. Could you possibly upload it?
to:
https://skyline.ms/files.url
For the developer who owns this feature (Rita Chupalov) to have a closer look.
--Brendan
|
| |
| roman sakson responded: |
2021-07-12 11:33 |
Hi Brendan,
thank you, I uploaded it under my name and I am also attaching it here, since it is very small. I am not sure, however, would you also need the raw file? At least I cannot browse the Full-Scan spectra without having the raw data directory mapped...
Please, let me know.
Roman
|
|
| |
| Rita Chupalov responded: |
2021-07-12 13:27 |
Hi Roman,
would you mind uploading the .raw file as well. Otherwise I cannot really see the spectra since spectra data is not part of the document itself.
Thank you,
Rita
|
| |
| roman sakson responded: |
2021-07-12 13:45 |
Hi Rita,
sure, the file's name is qExHF01_13483.raw, "From Roman for Rita".
Thank you for looking into it!
Roman
|
| |
| Rita Chupalov responded: |
2021-07-13 15:55 |
Hi Roman,
thanks for sharing your data file. I cannot reproduce your problem yet, though. When I click on the "Show rank and type" button it produces correctly annotated spectrum. Sometimes the annotations can appear when you vertically zoom the spectrum. Is it the case for you?
Could you open the Help/About dialog and send me the exact version/build number? I assume you are using 21.1, but I would like to make sure I use the same codebase.
~Rita
|
| |
| roman sakson responded: |
2021-07-14 11:08 |
Hi Rita,
I was, indeed, using 21.1.0.146 (9b577b3b8). Thank you for pointing it out, I tested again on another computer and there it worked perfectly. I could not figure out the reason but a fresh Skyline installation solved the issue for me. I am sorry for the trouble!
However, now that I have this great new feature at my disposal, I would also like to create mirror spectra between my experimental MS/MS data and Prosit predictions in Skyline, as this would kind of allow me to do "visual ID" without actual spectral libraries in place. I would guess that there is no easy way to mirror the content of the Full Scan window and of the Spectral Library window? Could I somehow easily extract MS/MS information from my Thermo raw file with Skyline to do a pseudo-spectral library? I think something like that is enabled for the Sciex MIDAS workflow. I hope that this is not too confusing.
Thanks again for your support!
Roman
|
| |
| Rita Chupalov responded: |
2021-07-14 14:03 |
Hi Roman,
I'm glad it worked out for you and that you find this feature useful.
Yes, I think it is possible to create a library from your raw file, but to do it you need to generate a search result file using Sequest or Crux first. This will let Skyline know which spectra in your raw file belong to each peptide of interest.
After it is done, this tutorial explains the library building process.
Hope this helps,
Rita
|
| |
| Rita Chupalov responded: |
2021-07-14 14:15 |
|
| |
| roman sakson responded: |
2021-07-15 14:18 |
Hi Rita,
thank you for the links and the suggestions! Unfortunately, I don't think that the normal way to create a peptide match from DDA data would get me where I want, since I am dealing with some bad-behaved peptides that normally do not get an ID from algorithms like Sequest. However, Prosit is supposed to be able to predict such difficult MS/MS patterns at least to some extent. Therefore, I would very much like to mirror my raw MS/MS data (or data like mgf; mzXML or mzML without ID) to Prosit predictions directly. Is there any way to do that?
Thanks,
Roman
|
| |
| Rita Chupalov responded: |
2021-07-15 15:01 |
Hi Roman,
Unfortunately Skyline's Full Scan Viewer doesn't support mirroring at the moment.
I will suggest it to the team as a new feature to implement.
|
| |
| roman sakson responded: |
2021-07-15 15:19 |
|
| |
 I_Full_Scan_Conventional.PNG
I_Full_Scan_Conventional.PNG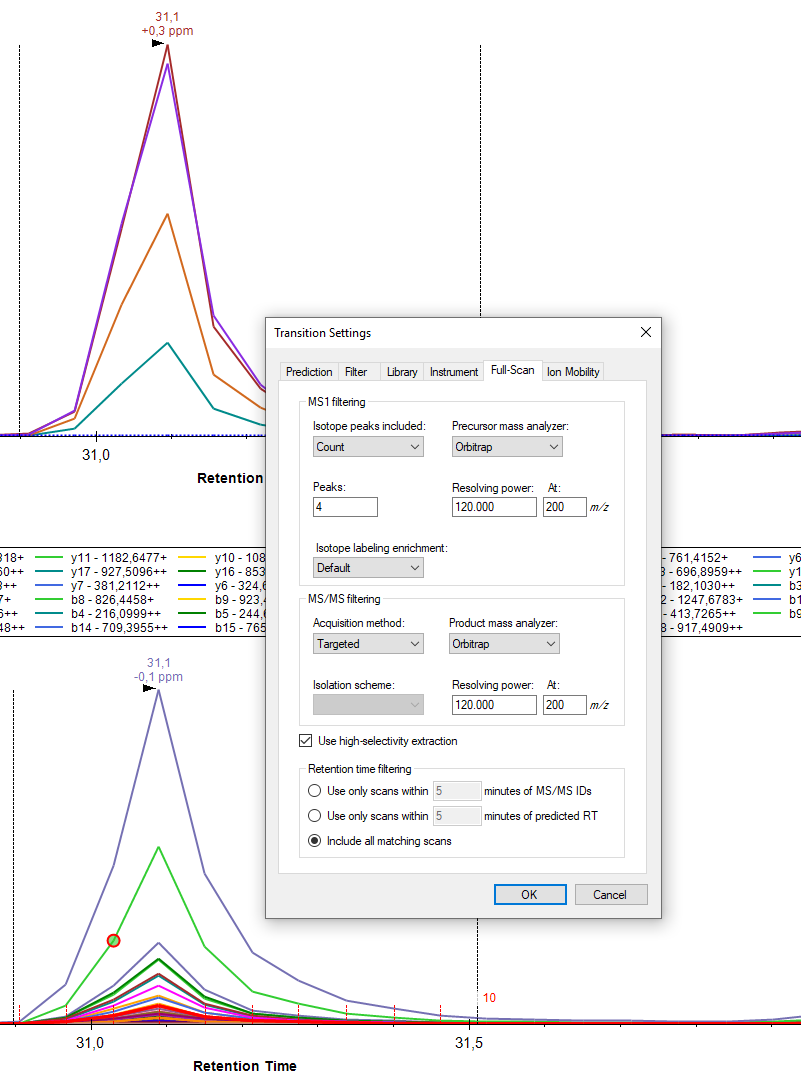 Full_Scan_Settings.PNG
Full_Scan_Settings.PNG