| no files detected during import of library from fragpipe | kguehrs | 2021-06-15 03:49 | |||||||||||||||||||||||||||||||||||||||||||||
Hello Skyline team, I tried to import MSFragger results into skyline. I followed the procedure given in tutorial on the following website by the develeopers of FragPipe. https://msfragger.nesvilab.org/tutorial_skyline.html I used the default method "DIA_Umpire_speclib" and the analysis by Fragpipe was successful. I can see the expected number of mzml, pepxml, calibrated.mgf, and interact....pep.xml files in the MSFragger subdirectory of the directory that also contains the raw files. This subdirectory also contains the protein.fas and tsv files generated by MSFragger. The import of the interact...pep.xml files however failed with the error message shown in the attached file. For me, all the files described in the nesvilab tutorial are available and assume that here is either some file structure problem or some more syntax problem that I do not understand. Best Karl-Heinz |
|||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
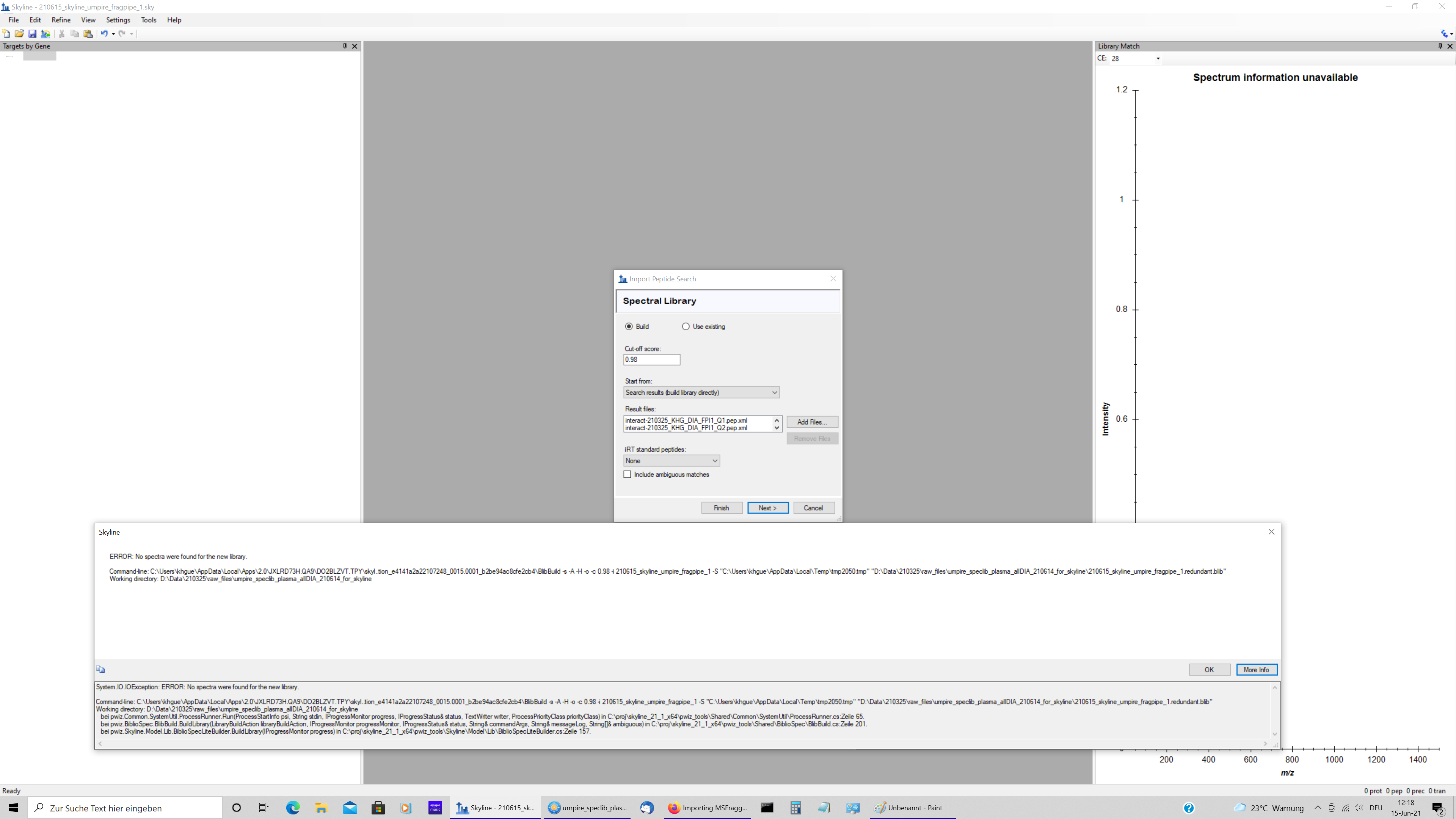 skyline_error.png
skyline_error.png