Can you send us your Skyline document and one of your raw files? I can help you figure out the easiest way to get past this problem.
In Skyline, you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
You can upload that .zip file, and one of your .raw files here:
https://skyline.ms/files.url
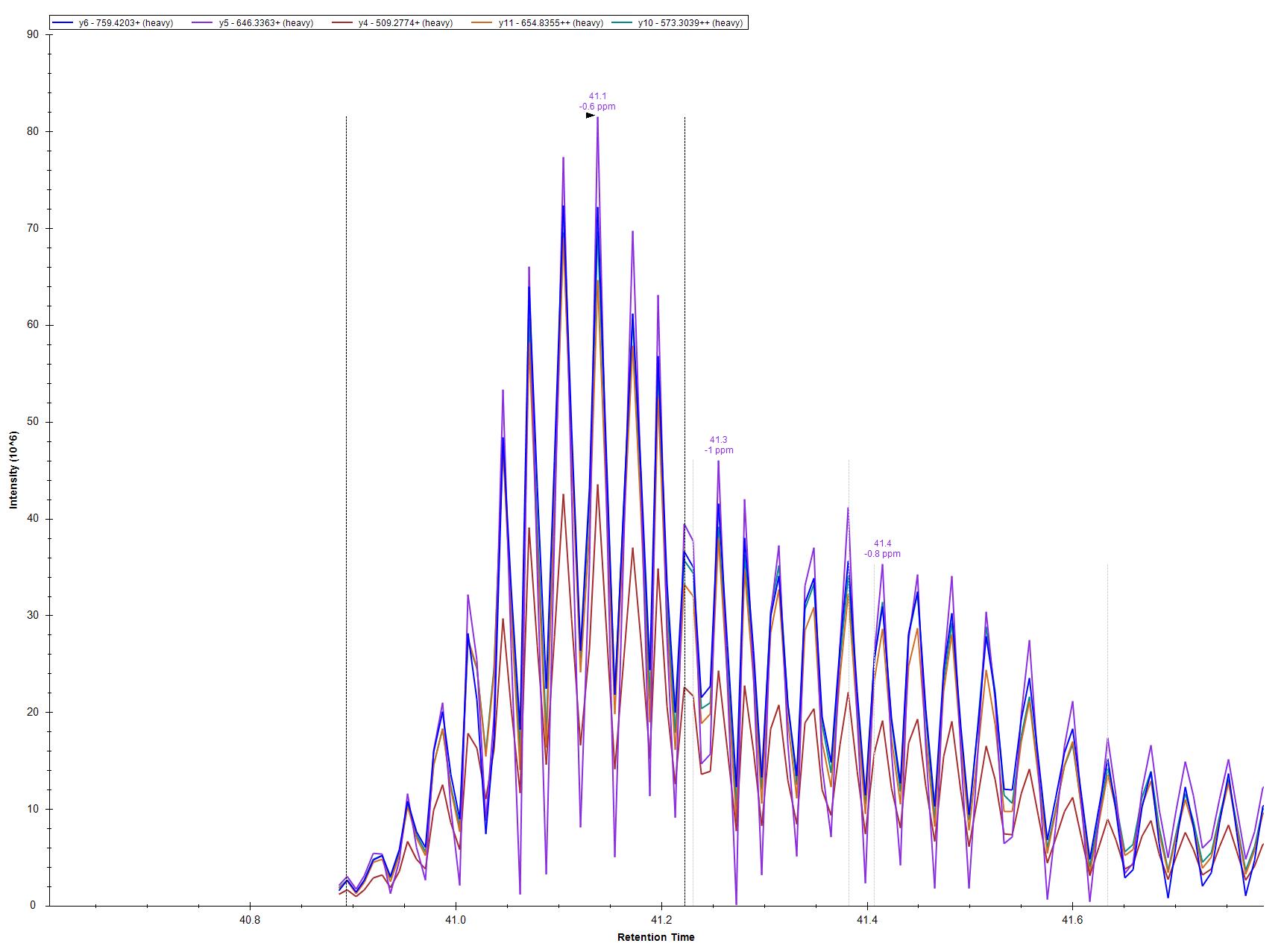
The usual reason that chromatograms look jagged like that is that they are including two different types of MS2 scans.
This can happen with SureQuant methods when a heavy precursor has a m/z which is very close to a light precursor belonging to a different peptide.
Skyline decides which MS2 spectra should be used for which precursors based on the "MS/MS filtering Acquisition Method" and "Isolation Scheme" settings at:
Settings > Transition Settings > Full Scan
Sometimes, it is possible to customize the isolation scheme so that Skyline never gets confused about whether a particular MS2 spectrum belongs to a particular heavy or a different light precursor.
However, if your SureQuant method is targeting a large number of peptides, there will always be some cases where you have two peptides whose m/z's are so close to each other that there is no way to use the Isolation Scheme to tell Skyline how to distinguish them.
In these cases, there is a different way to tell Skyline which spectrum goes with which peptide. Your mass spectrometer method needs to add a "Scan Description" property to each spectrum that will tell Skyline which peptide the spectrum was for. The rules for exactly what that "Scan Description" property need to look like are complicated, and I am not sure whether you need to know what those rules are, or whether that has already been taken care of in the instrument method that you got from Thermo.
In case, you need to know what the rules are for what the Scan Description is supposed to be, those rules are:
The Scan Description property needs to start with either "SQ_ENDO_" (for light peptides), or "SQ_IS_" (for heavy peptides). After that, it should have either "K_" or "R_" depending on which amino acid is labeled, followed by the mass difference of the label. Then, it ends with the charge state of the precursor.
Hear are some example Scan Description property values.
SQ_IS_R+10_2
SQ_ENDO_K+8_2
Skyline only pays attention to the Scan Description property if "Triggered Acquisition" is checked at "Settings > Transition Settings > Instrument".
Anyway, please send us your Skyline document and one of your raw mass spectrometry data files and we can help you find the best way to fix this problem.
-- Nick
 graph.jpg
graph.jpg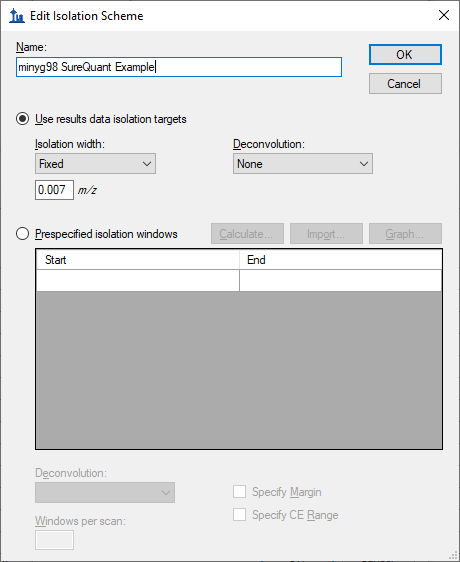 IsolationScheme.png
IsolationScheme.png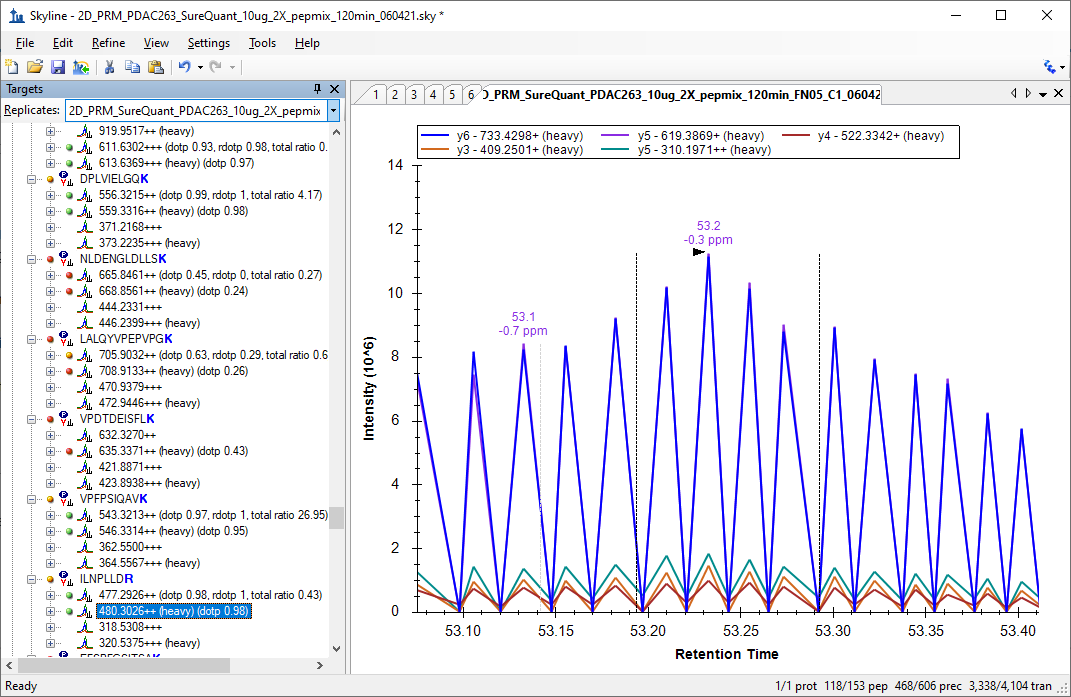 ProblematicPeptide.png
ProblematicPeptide.png