| iRT peak pickinpicking wrong peak of some replicates) | boycea | 2021-05-03 19:58 | |||||||||||||||||
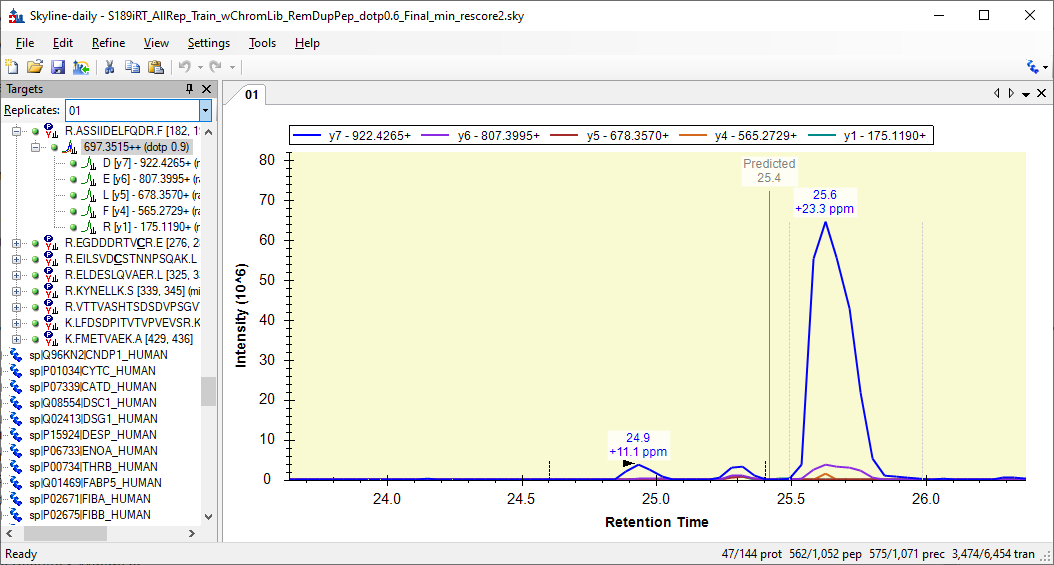
After setting up our iRT predicted calculator, chrom libraries, replicates uploaded, and reintegrating by the mProphey peak scoring model and 0.01 significant q values, we noticed that some peptides don't have the same, nor correct peaks picked automatically among all their replicates. On top of that, those incorrect peaks had higher ppm (>10), lower dotp (<0.7), and further from the predicted time than other peaks that seems like they should be automatically choosing (on predicted RT, dotp=0.99, ppm<1). Is there a filter or refinement in skyline that can automatically pick the peaks that seem more likely based on certain dotp, ppm, and RT thresholds, and consistently across all replicates of all peptides? Also, more specifically, is there a filter for eliminating or reintegrating peptides that don't have peaks within a certain ppm threshold. I have a document with a couple of examples, if needed. In the example provided, for gene CLU, peptide ASSII, most replicates chosen is the small peak in the middle of 3 at RT ~25.3, dotp >0.98, and ppm<10. However, other replicates (e.g. 1, 13, 27), with less ideal ppm, dotp, and RT peaks (the one on the right, ~RT 25.5) were chosen, and when we manually integrated the middle peak in the those replicates, it gave us favorable ppm and dotp like the rest. Is there further refinement we can so that more peptides will have more consistent peaks chosen among all the replicates without having to go through each of them? Thanks! Aaron |
|||||||||||||||||||
| |||||||||||||||||||
 RescoredPeak.png
RescoredPeak.png