| Nick Shulman responded: |
2021-03-31 09:47 |
Can you send me your Skyline Document .sky file?
(Normally, I would ask you to create a .sky.zip file, but since you say you cannot open the file, you probably also cannot use the "File > Share" menu item to create a .sky.zip file).
What error are you seeing when you try to open the document? Are you seeing an error with a message that looks like "error reading mz values"? That sort of error is always caused by a bug in Skyline, where we are not being consistent in how the m/z of a precursor or transition gets calculated. (Sometimes this error shows itself when you upgrade to a newer version of Skyline where we have fixed some bug that happened in the past, and sometimes this happens because Skyline did not recalculate the m/z of something when the settings changed).
When you send me your .sky file, I will probably be able to figure out what is going wrong?
By the way, are you using the Crosslinked Peptide feature that was added in Skyline 20.2? That feature allows you to designate that a modification is a crosslinker, and then you can use the Modify Peptide dialog to specify that the crosslinker is a looplink or links to a different peptide. We have made some great improvements to the Crosslinking feature in Skyline-Daily, which makes it much easier to define quite complicated crosslinked peptides.
If your .sky file is less than 50MB you can attach it to this support request.
Otherwise, you can upload it here:
https://skyline.ms/files.url
-- Nick |
| |
| jmr responded: |
2021-03-31 13:23 |
See attached power point and skyfiles. Error is in the powerpoint.
Not sure if I am using the Crosslinked Peptide feature and if I am, I do not think I am doing it correctly. Can you send me instructions? |
|
| |
| Nick Shulman responded: |
2021-03-31 14:02 |
Thanks for sending those files.
There are a couple of bugs here:
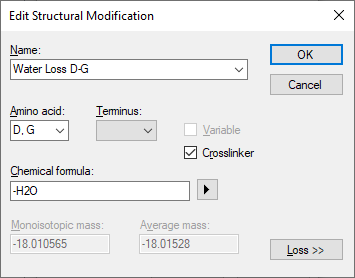
1. Skyline should not be giving you an error about "Explicit Loss Only Modification" when you open your document. I will fix this, but you can work around this by defining your crosslink modification differently. The "-H2O" is the actual formula of the modification, not a neutral loss (see attached image). A neutral loss would be a chemical change to fragment ions, that is not found on the intact crosslinked peptide. If you define your modification that way, you will not get the bogus error that you are getting when you open your Skyline document.
2. Before you hit the OK button on the "Edit Linked Peptides" dialog, you should put the focus on one of the other text boxes on the form. For some reason, Skyline is getting confused by the fact that the focus is in the bottom row of the "Linked Peptides" grid and thinks you are trying to add a new peptide to this list.
I will fix both of these bugs, but it looks like it should be straightforward for you to work around them.
Let me know if you have any other problems, or other questions.
-- Nick |
|
| |
| jmr responded: |
2021-03-31 14:48 |
Thanks so much! This worked.
Also wondering why I am not able to see my isotopic distribution for product ions when I look at replicate comparison. Is this a setting?
When I look at peak area peptide comparison, I can see the various fragment ions, but not as a stack plot , like I would prefer. |
|
| |
| Nick Shulman responded: |
2021-03-31 22:13 |
I am not sure that I understand your new question(s).
Are you asking whether Skyline can extract multiple chromatograms corresponding to different isotopes for fragment ions? Skyline only extracts the chromatogram corresponding to the monoisotopic mass of fragment ions. Skyline extracts chromatograms for the full isotope envelope only for precursor (MS1) chromatograms.
I don't believe there is any way to get the Peak Area Peptide Comparison to show any sort of stacked bar chart.
-- Nick |
| |
| jmr responded: |
2021-04-01 07:20 |
OK. I was wonder why the bottom 1/2 of the Peak Areas (replicate comp) did not show a bar graph but the precursor did. I can see the bar graph when I select Peak Areas - Peptide comp but it isn't stacked and I cannot show the legend. I am not really comparing anything. I just want to see the ion distribution of fragments in a stack instead of side by side. It has been a while since I have used skyline and I know I used to do this, but maybe I exported each fragment's peak area and created the plot in excel.
Also - can you tell me if my settings are correct for Transition settings-Full Scan? I am importing a raw file from an LTQXL-Orbitrap. I am running a DDA method where scan event 1 is Full Scan in Orbi using 7500 or 15000 resolution, scan event 2 is MSMS in IT, scan event 3 is MS3 in IT. See attached file.
Thanks for your help. |
|
| |
| jmr responded: |
2021-04-01 07:40 |
I now imported other files and no matter what settings I use, I cannot see the bar graph for the products. Am I doing something wrong? See attached. |
|
| |
| Nick Shulman responded: |
2021-04-01 08:00 |
I see that the Transitions are being drawn with a hatch (tiny checkerboard) pattern. That indicates that the transitions are being treated as non-quantitative. When your Transition Full Scan MS/MS Acquisition Method is "DDA", then all MS2 transitions are treated as non-quantitative. Non-quantitative transitions are used in peak finding, but their peak areas are excluded from calculated values such as "Total Area".
You can also choose to mark particular transitions as non-quantitative either by right-clicking on them in the Targets tree or by setting values in the "Quantitative" column in the Document Grid.
What you are seeing looks like a bug in Skyline. I am not sure exactly what non-quantitative transitions are supposed to look like in the peak area bar graph, but I am pretty sure they should not look like what you are seeing.
It would be very helpful if you could send me your Skyline document.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file is less than 50MB you can attach it to this support request. Otherwise, you can upload it here:
https://skyline.ms/files.url
-- Nick |
| |
| jmr responded: |
2021-04-01 08:43 |
Thank you Nick. Is this a secure site; my sequence is confidential. |
| |
| jmr responded: |
2021-04-01 09:29 |
I actually see that the url is not secure. I can email you a sharefile link or can you send one to me and I will send you my skyline file. Let me know your preference for sharing a file with high security.
Thanks again.
Jeanne |
| |
| Nick Shulman responded: |
2021-04-01 10:09 |
I am able to reproduce the problem that you are seeing.
Whenever I have the Acquisition Method set to "DDA", the product ion part of the peak area graph ends up being blank. This is not the correct behavior. The product ions should only disappear if you check the menu item "View > Transitions > Only Quantitative".
I will try to fix this soon.
I will send you an email in case you ever have other confidential files you need to send us.
-- Nick |
| |
| jmr responded: |
2021-04-01 11:00 |
Great - Thanks! |
| |
 CrosslinkModification.png
CrosslinkModification.png loop example.PNG
loop example.PNG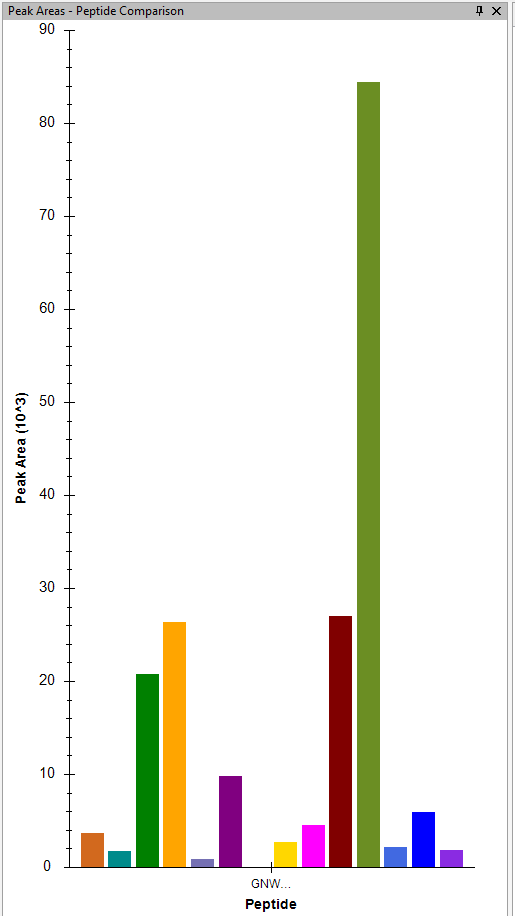 loop example-peptide comp.PNG
loop example-peptide comp.PNG Transition Settings-full scan.PNG
Transition Settings-full scan.PNG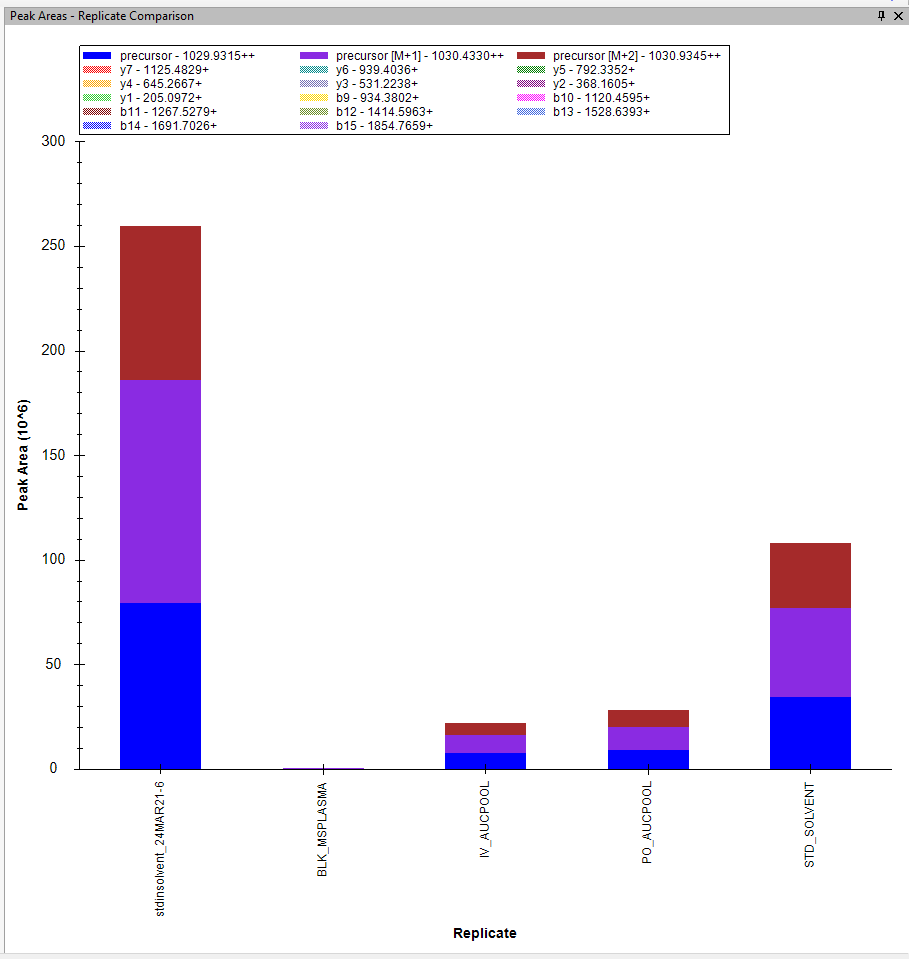 Transitions_All_no split graph.PNG
Transitions_All_no split graph.PNG Transitions_All_with split graph.PNG
Transitions_All_with split graph.PNG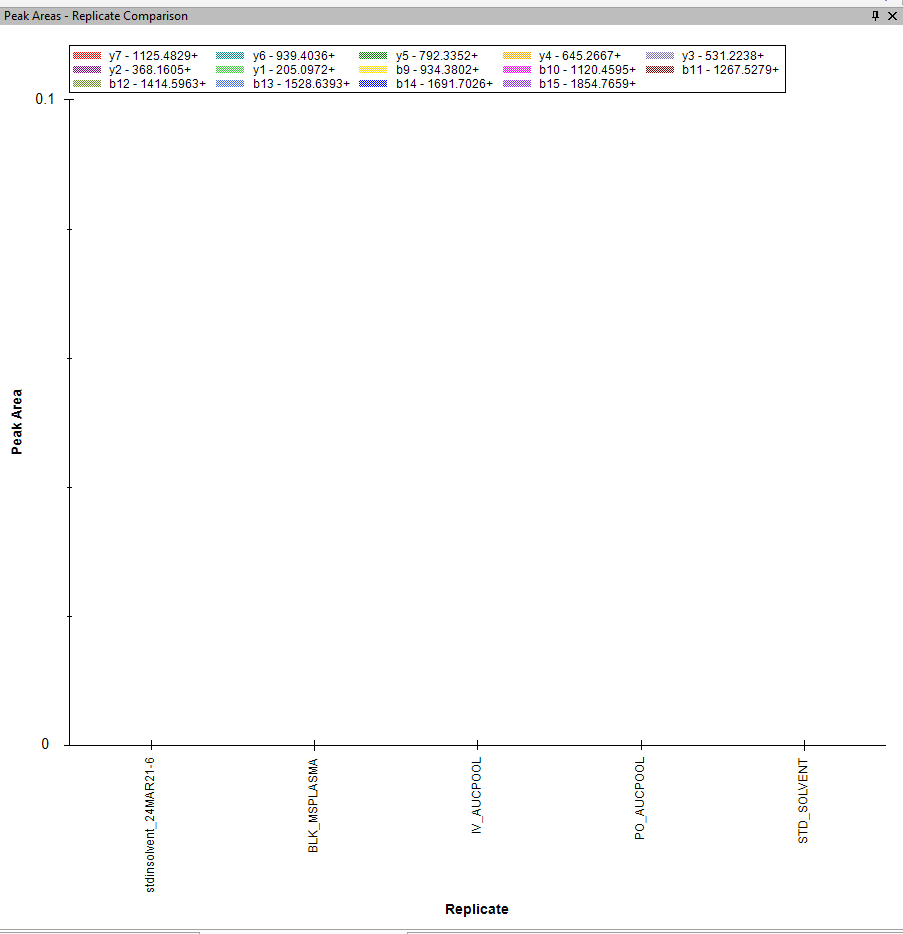 Transitions_products_no split graph.PNG
Transitions_products_no split graph.PNG