| Nick Shulman responded: |
2021-03-23 10:44 |
You can tell Skyline that an isotope modification will have a different retention time than the unmodified peptide.
To change that, edit the isotope modification by going to:
Settings > Peptide Settings > Modifications > Edit List > Edit
At the bottom of the "Edit Isotope Modification" dialog, there is a setting "Relative Retention Time". This defaults to "Matching". I think if you change it to "Preceding", I think Skyline might make it easier for you to adjust your peak boundaries the way you want them. (I actually am not sure exactly what happens when you choose "Preceding". I know that if you choose "Unknown", Skyline will let you adjust the peak boundaries of your precursors completely independently from each other, but I'm not sure what happens with "Preceding").
By the way, you can always adjust the peak boundaries of transitions independently of each other, but Skyline intentionally makes it difficult. If you right-click on the chromatogram and choose "Transitions > Single", then, if you have a single transition selected in the Targets tree, any adjustments that you make to peak boundaries will only be applied to that one transition.
-- Nick |
| |
| Brendan MacLean responded: |
2021-03-23 11:00 |
I was going to explain what Nick explained for peptides, but thanks to Nick, I don't have to. There is no equivalent for small molecules that I know of. However, you can always define your standard as a separate molecule and then Skyline will integrate them separately, and by using multiple selection in the Targets view (Shift-click or Ctrl-click) you can get Skyline to plot the two values on the same graphs. It is not quite as convenient as the way we group precursors for the same molecule, but it could suffice as a workaround. It would certainly allow you to get the peak integration boundaries and peak areas you are trying to achieve. If your goal is to get those into a report for statistical processing outside Skyline, that might be enough.
I would expect MS1 chromatograms to work better than PRM in a simple solution. With MS1, you are generally working against other ions, either because they are much more abundant than what you are trying to measure and fill the trap, reducing the precision with which your target molecule can be measured, or by direct interference with the molecule you are trying to measure. You could use SIM to solve the first issue, but only PRM can help you with interference and noise reduction.
We do hear from our pharma partners that they prefer MS1 for low complexity samples. Conversely, ion filtering with a quadrupole or FAIMS device appears to be much more important in high complexity and high dynamic range samples like cell lysates or plasma.
Thanks for posting to the Skyline support board.
--Brendan |
| |
| alejandro.cohen responded: |
2021-03-23 11:12 |
Thanks Brendan, Nick
I was about to mention the absence of that setting for small molecules (maybe a suggestion for future releases?).
With respect to Brendan's suggestion (ctrl-click), would the normalization still work? That is, will Skyline know one is the Internal Standard of the other automatically, for calibration curves and interpolating Values of unknowns?
Thanks again, Skyline rocks... I'm creating a small Skyline-sect here at Dalhousie University.
Best
Alejandro |
| |
| Brendan MacLean responded: |
2021-03-23 15:49 |
You would not get the automatic light:heavy normalization, but if you had a small enough number of these, you could use the "surrogate standard" mechanism to pair them up manually. Not great. Yes, it seems like an option like the retention time matching for peptides would be useful for small molecules. We just hadn't seen a case of that yet.
Thanks for the feedback.
--Brendan |
| |
| alejandro.cohen responded: |
2021-03-24 05:42 |
Thanks Brendan!
I have 18 targets. I created 36 molecules, just as you suggested. I do have some questions:
I did go through the tutorials, I created the Normalization Method Column, and selected ratio to Heavy, but can't find a way to individually link each molecule to it's respective surrogate (I would have imagined another column with a drop down menu linking each molecule to it's surrogate).
The tutorials only give an example of one surrogate.
I cant see the normalized areas, so clearly I'm screwing up.
Attached the file for you to inspect, if you have the time to inspect it. |
| |
| Nick Shulman responded: |
2021-03-24 07:20 |
|
| |
| alejandro.cohen responded: |
2021-03-24 09:00 |
WORKED!!! Solved. Thanks N&B! |
| |
| alejandro.cohen responded: |
2021-11-26 07:29 |
Nick, Brendan,
I'm following up on this thread. Just a few notes:
-Request feature: I'm just being repetitive: It would be fantastic if for small molecules you could set different retention times for light and heavy (deuterated in my case) within the same molecule. For highly labelled targets (say 9 deuteriums atoms), the Rt difference is surprisingly high.
I'm sure it's lots of coding, but having the ability to do this avoids the extra time of your suggestion above (Two molecule list names, and surrogate Internal standard normalization.), which is momentarily working well.
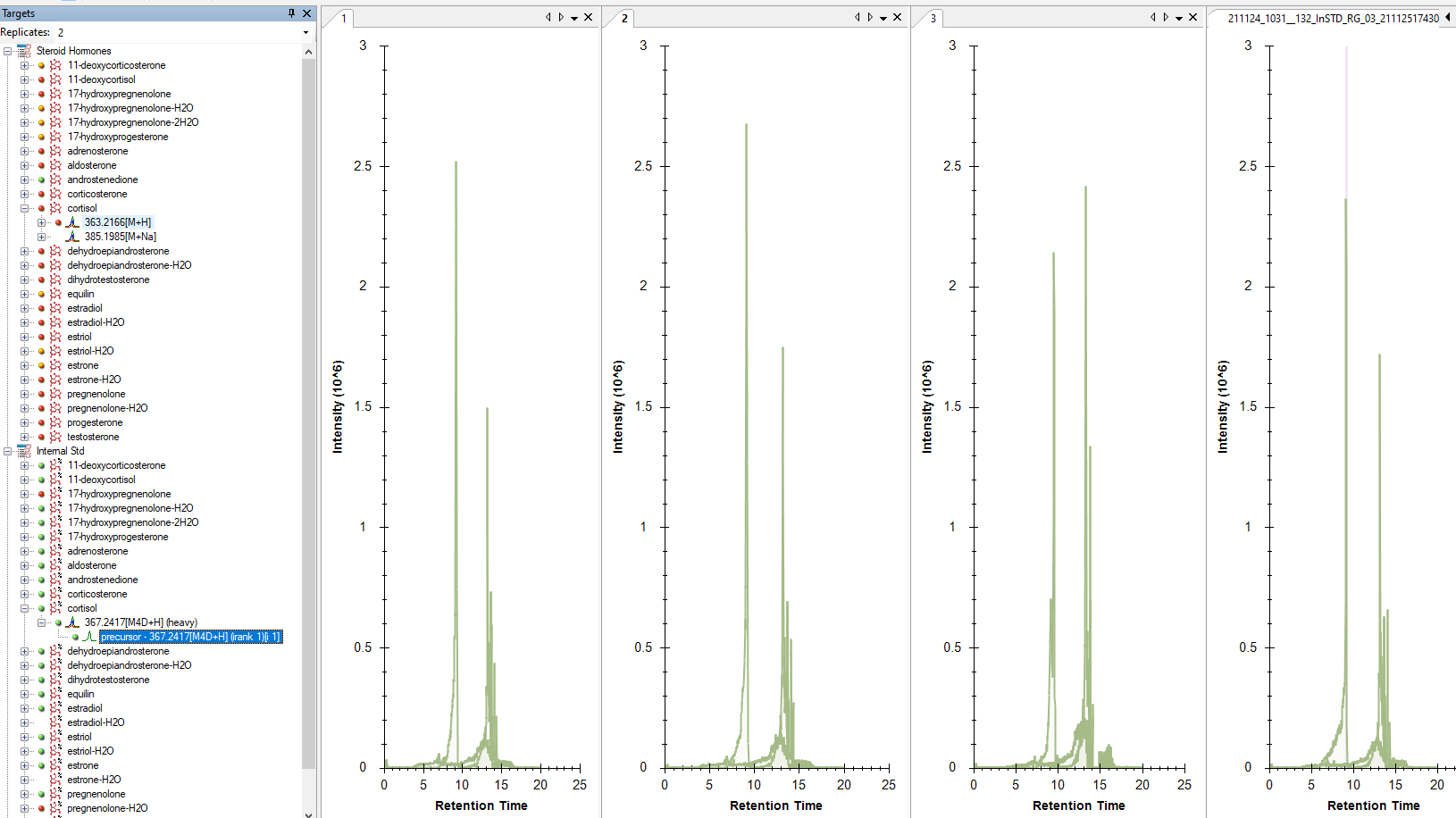
On this same issue, I've followed Brendan's advice: "and by using multiple selection in the Targets view (Shift-click or Ctrl-click) you can get Skyline to plot the two values on the same graphs"
I've attached a screen capture of this. The fact the chromatogram colors are identical for selected precursors AND that there is no legend makes this option not very useful for inspecting the data.
Cheers!!!
Alejandro |
|
| |
| alejandro.cohen responded: |
2021-11-26 13:33 |
Quick follow-up
I think I know what the problem is with the : "and by using multiple selection in the Targets view (Shift-click or Ctrl-click) you can get Skyline to plot the two values on the same graphs"
My internal standards in their Molecule List are named identically to the light ones. IF If rename them with a suffix or prefix, they DO appear with different color (and legend) with I select multiple targets with the Shift-click trick :)
Thanks!!! |
| |
 Screenshot 2021-11-26 112403.png
Screenshot 2021-11-26 112403.png