Can you send us your files? That would be your Skyline document, msms.txt, and if you have them mqpar.xml and modifications.xml.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document.
In addition to that .zip file, you should also send us your msms.txt, mqpar.xml (if you have one) and modifications.xml.
If those files are less than 50MB you can attach them to this support request.
Otherwise, you can upload them here:
https://skyline.ms/files.url
If "Import Peptide Search" is not working for you, you might have better luck doing the steps separately.
The first thing that Import Peptide Search does is build the library, which you can get to by going to:
Settings > Peptide Settings > Libraries > Build...
The next thing that Import Peptide Search does is the equivalent of "File > Import > FASTA". However, if you are doing the steps yourself, you might instead find it easier to go to:
View > Spectral Libraries > Add All
Adding peptides from the spectral library viewer instead of by importing a fasta file sometimes solves problems for people, but I imagine you will still be seeing the warnings you are seeing about some modifications not being interpreted.
We will probably be able to figure out what is going wrong if we see your Skyline document and peptide search results.
-- Nick
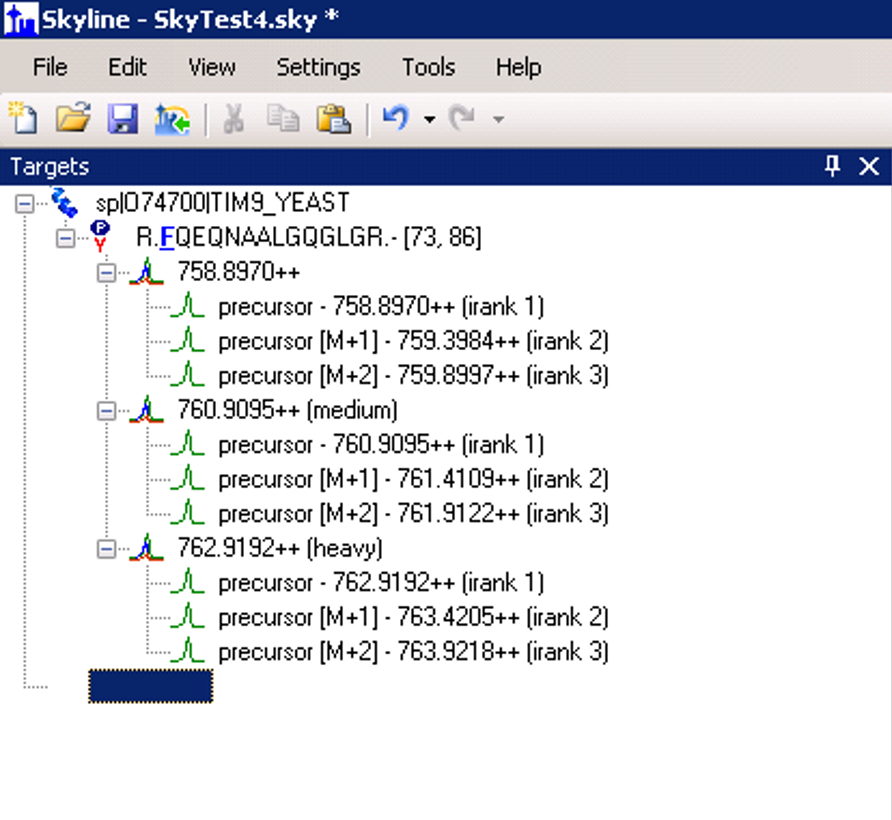 Settings.png
Settings.png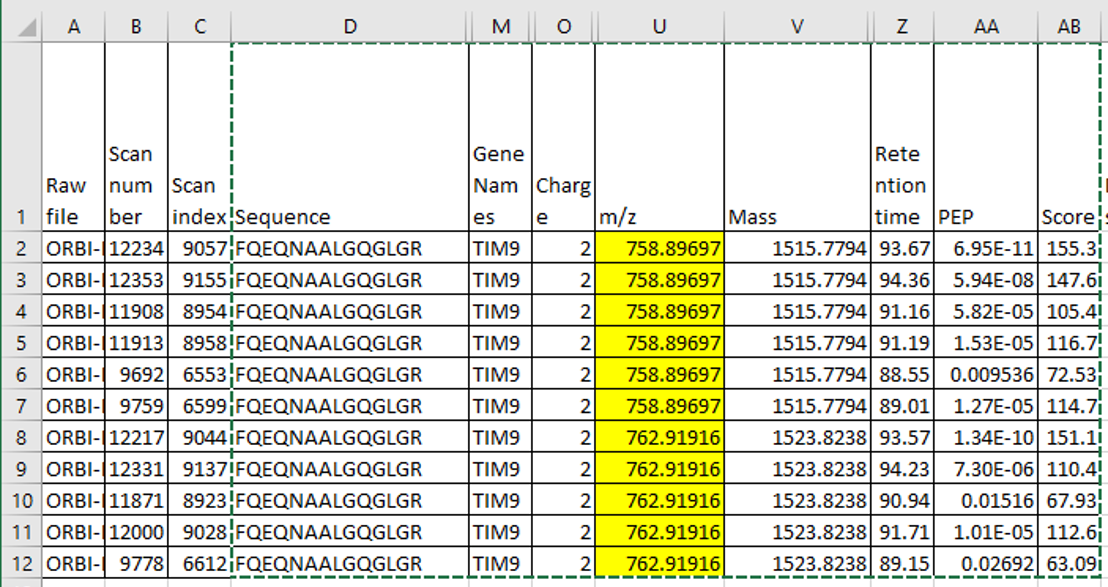 msmsScreenshot.png
msmsScreenshot.png MaxQuantEvidence.png
MaxQuantEvidence.png