I think Skyline might have been confused by the fact that your 1st_1tran__0003.csv only has 6 steps for each compound (-2 to +3), instead of the usual 7.
So your transition list looks like this:
|Compound|Retention Time (min)|RT Window (min)|Polarity|Precursor (m/z)|Product (m/z)|Collision Energy (V)|
|-------|--------|----------|------------|-----------|-----------|------------|----------|
|S6b_T.Cathine/[Mo_S6]Phenylpropanolamine(+1).-2|1.95|1.2|Positive|152.1|117.08|6|
|S6b_T.Cathine/[Mo_S6]Phenylpropanolamine(+1).-1|1.95|1.2|Positive|152.1|117.09|12|
|S6b_T.Cathine/[Mo_S6]Phenylpropanolamine(+1)|1.95|1.2|Positive|152.1|117.1|18|
|S6b_T.Cathine/[Mo_S6]Phenylpropanolamine(+1).1|1.95|1.2|Positive|152.1|117.11|24|
|S6b_T.Cathine/[Mo_S6]Phenylpropanolamine(+1).2|1.95|1.2|Positive|152.1|117.12|30|
|S6b_T.Cathine/[Mo_S6]Phenylpropanolamine(+1).3|1.95|1.2|Positive|152.1|117.13|36|
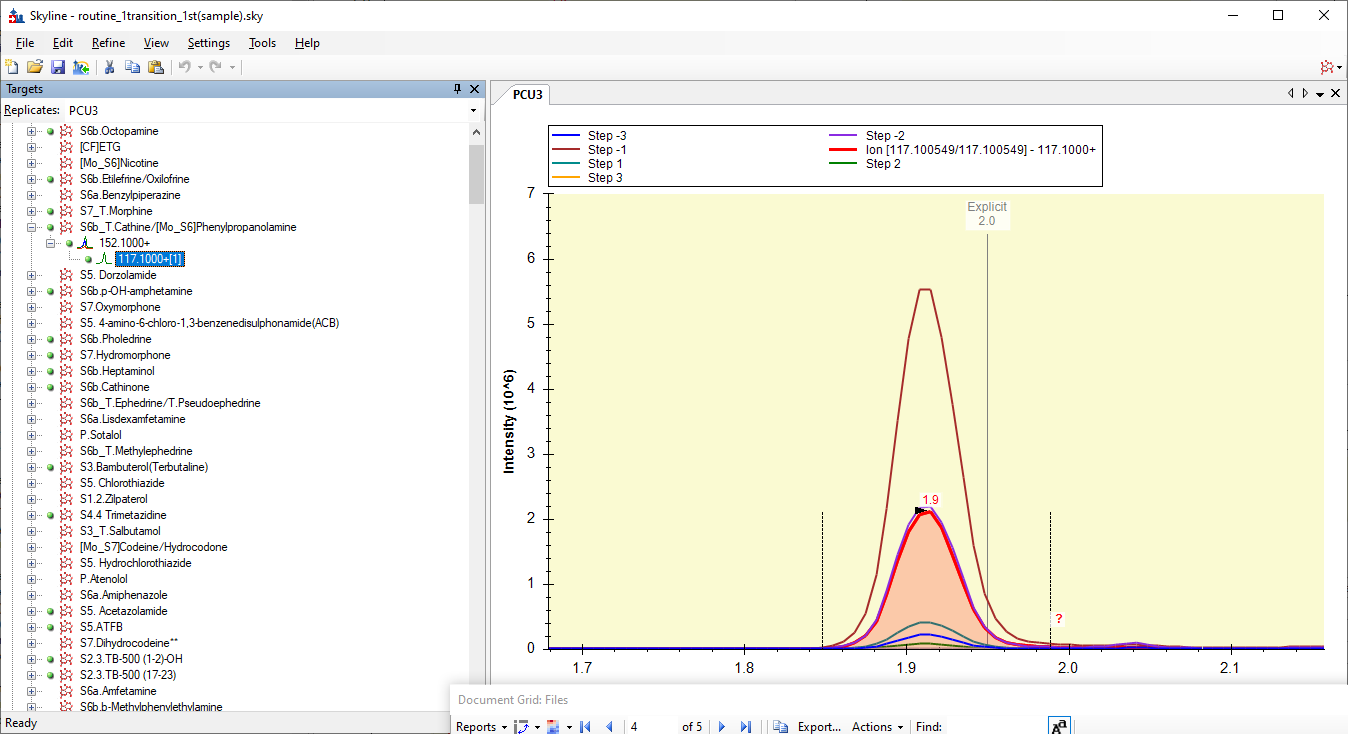
However, when Skyline extracts chromatograms from that, Skyline does not know that it was supposed to start a -2 and go to +3. Instead, Skyline thinks it went from -3 to +2, and the +3 chromatogram is missing.
So, the chromatograms in Skyline look like the attached screenshot.
It looks like Skyline is showing you all the chromatograms from step -3 to +3, but the yellow +3 chromatogram is actually missing.
Skyline ends up thinking that Step -1 is the best chromatogram, and so, when Skyline exports your transition list for the second round of optimization, the CE values that Skyline exports are all centered around 12, because that's what Skyline though yielded the biggest chromatogram.
However, in reality, since Skyline was confused about which chromatogram had which collision energy, the best chromatogram had a collision energy of 18.
The thing to keep in mind about Skyline and CE optimization is that Skyline has no idea what the collision energies in your raw file are. Skyline notices a set of chromatogams with identical Q1 values and where the Q3 number differs by 0.01, and then Skyline has to make assumptions about what their collision energies were.
-- Nick
 CE_Optimization_Run.png
CE_Optimization_Run.png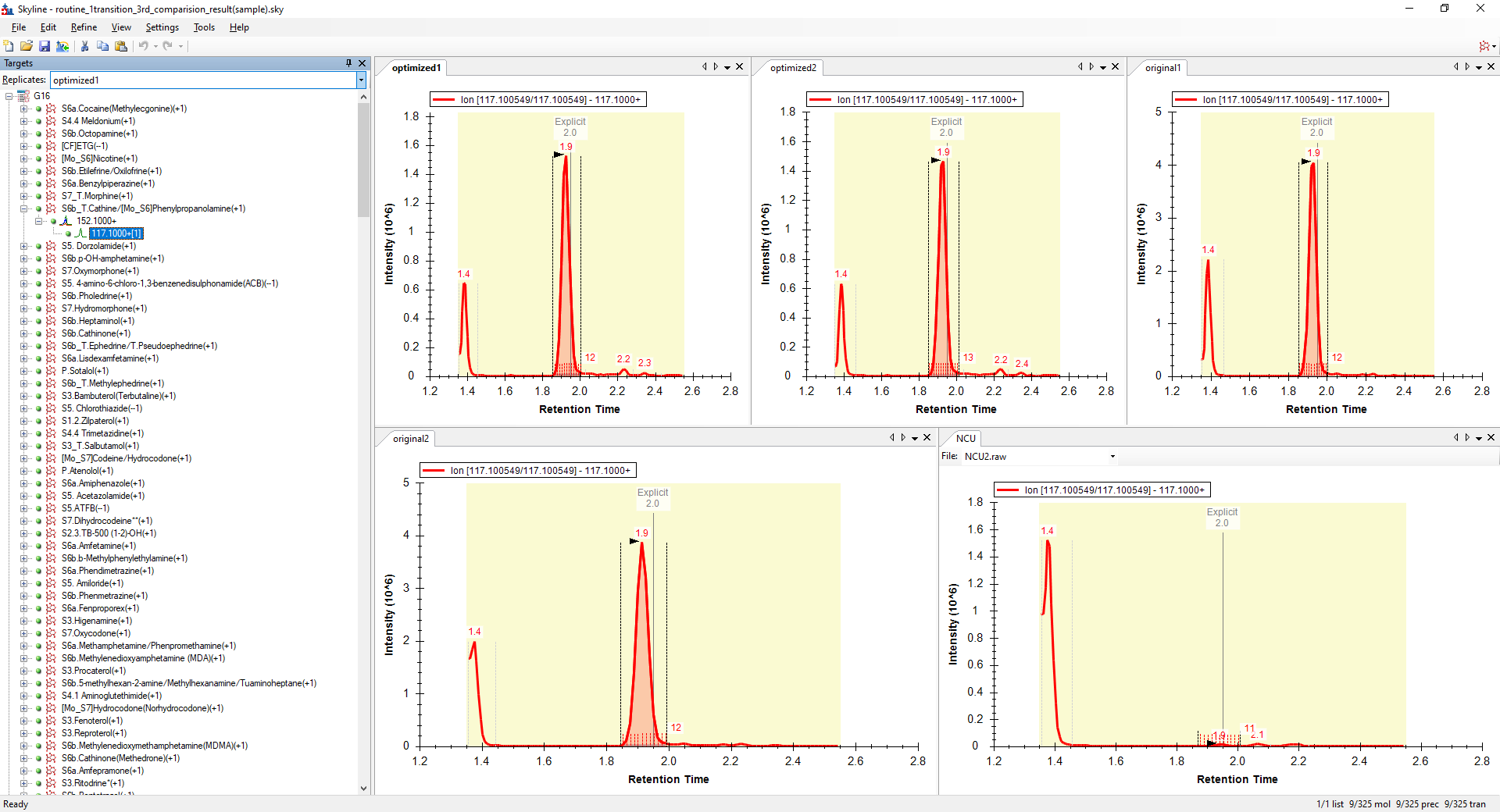 Comparison.png
Comparison.png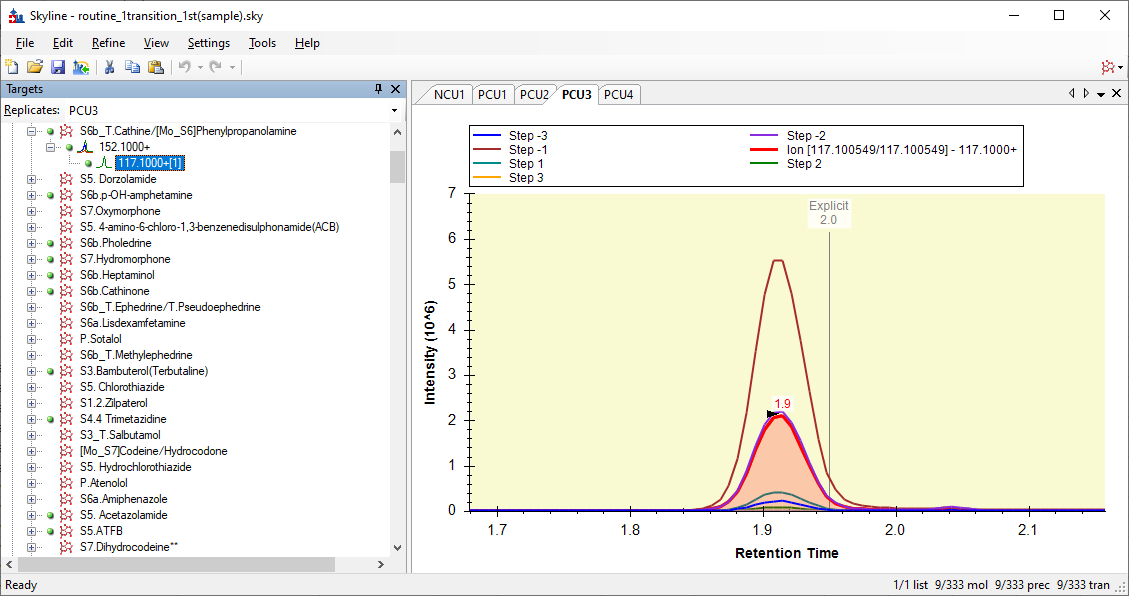 OptStepChromatograms.png
OptStepChromatograms.png