| |
| Nick Shulman responded: |
2021-01-20 07:48 |
Xin,
Can you send us your Skyline document and raw file?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing the Skyline document and supporting files including extracted chromatograms.
You should also send us your raw file, which for Waters I think means Zip up the .raw directory.
If those files are less than 50MB you can attach them to this support request. Otherwise, you can upload them here:
https://skyline.ms/files.url
I am not sure that I can tell what is going on from your screenshot. I see that there is a straight line in the chromatogram between 2.8 and 3.5 minutes. Usually a straight line like that indicates that there was a time when there were no MS2 scans which matched the peptide. Sometimes this can be fixed by changing the MS/MS Acquisition Method at "Settings > Transition Settings > Full Scan", or sometimes the "Method Match Tolerance" at "Settings > Transition Settings > Instrument".
If you want to know what spectra Skyline is able to see in your raw file, I recommend that you look at the file using SeeMS.exe which comes with ProteoWizard. You can install ProteoWizard from here:
http://proteowizard.sourceforge.net/download.html
Skyline is able to see all of the same spectra in your raw file as SeeMS.exe. Skyline uses the information in the "Isolation Windows" column to decide which spectra should contribute to which peptide's chromatogram.
-- Nick |
| |
| xin huang responded: |
2021-01-20 08:33 |
Dear Nick,
Thanks for your prompt reply, please see the attached compressed skyline file and the compressed raw. file. Please let me know if you need more info and what has happened in there, I will download ProteoWizard to have a look as well.
Best,
Xin |
|
| |
| Nick Shulman responded: |
2021-01-20 09:51 |
Xin,
In Skyline, you should go to:
Settings > Transition Settings > Full Scan
and change the MS/MS filtering Acquisition Method to "Targeted".
Then, you should reimport your chromatograms by going to:
Edit > Manage Results > Reimport
When you do that, your precursor and product ion chromatograms for that peptide will look like the picture that I have attached.
Currently, you have your Acquisition Method set to "DIA", and you are using the Isolation scheme "All Ions".
The isolation scheme "All Ions" should be used in mass spectrometers that really are not doing any selection on the ions before fragmenting them. In your experiment, you really did isolate particular m/z's before fragmenting them.
Usually you could also use the "Results Only" isolation scheme, except in your case, the raw file does not have any information about how wide the isolation windows were. Skyline can only tell which mass was at the center of each isolation window. If you want to use "DIA" as your isolation method, you would also need to prespecify all of the isolation windows.
I actually do not know why you are seeing the behavior that you are seeing where when your isolation scheme is "All Ions", your chromatogram ends up missing the exact points that you wanted it to have. I might need to look into that.
Anyway, I think all your problems will be fixed if you change your MS/MS Acquisition Method to "Targeted".
-- Nick |
|
| |
| Nick Shulman responded: |
2021-01-20 12:04 |
Xin,
It turns out that when your Acquisition Method is "All Ions", Skyline will skip over all spectra in a Waters file whose Function Number is 3 or greater.
If you change the Acquisition Method to something else, like "Targeted", then Skyline will still skip over spectra whose Function Number is equal to 3, but will include spectra whose Function Number is greater than 3.
-- Nick |
| |
| Brian Pratt responded: |
2021-01-20 12:26 |
By way of explanation: function 3 is where the lockmass scans are usually found. |
| |
| xin huang responded: |
2021-01-21 02:04 |
Hi Nick and Brian,
Thanks a lot, change of the MS/MS acquisition method to 'targeted' did solve my problem for this data.
I still don't fully understand the logic behind the Function number. My Masslynx only shows five functions, Function 1 is always MS data, Function 2 and 3 are MS/MS data (no matter how many MRMs I have at the same time, 2 or 3 or 5 MRMs, Masslynx always try to squeeze them into these 2 Functions, so sometimes if there are overlapping time window, I don't see the overlapped ones in Masslynx), lockmass in Function 4, and UV in Function 5. When I open the fdc. file, I see the 5 MRM are actually in Function number 2,3,4,5,2., So earlier I was playing with the fdc. file and change the Function numbers manually.
I just tried to import this time window overlapped data with 5 MRMs to Skyline with both 'targeted' acquisition and 'DIA and all ions', and obtained very different chromatograms. Could you have a look to help me understand this? When I have 5 MRM light peptides detection, and another 5 MRM heavy labelled peptides for quantification, there will be overlapping time windows, and I wonder how the data acquisition goes.
Best,
Xin |
|
| |
| Nick Shulman responded: |
2021-01-21 05:59 |
Xin,
I am not sure what you are asking now. Can you send us a screenshot of what you are looking at?
By the way, it was incorrect when I said that Skyline would skip over spectra whose function number was equal to 3. Skyline has a more complicated way of deciding whether a spectrum is a lockmass spectrum (Brian is the expert on this) and when you set the acquisition method to Targeted, Skyline is not skipping over any of the spectra in your files.
When you set the isolation scheme to "All Ions", Skyline really does skip over all spectra whose function number is 3 or greater.
When a peptide has both MS1 and MS2 chromatograms, Skyline truncates the MS1 chromatograms so that they are the same length as the MS2 chromatograms. The reason that Skyline does this is because it is the correct thing to do when you have a scheduled PRM method. In your "All Ions" Skyline document, all of your peptides have MS2 chromatograms that go from 4.0 to 7.2 minutes. For this reason, all of those peptides have their MS1 chromatograms truncated to that time range.
In your Targeted Skyline document, some of your peptides do not have any MS2 chromatograms, because their precursor does not match anything in your raw file. For this reason, those peptides keep their full length MS1 chromatograms.
Is that the difference that you are asking about?
I do see that your peptide "RGVGPSVGV" is not getting any MS2 chromatograms. This is because the m/z of that peptide is 414.2403, and the spectra in your raw file were isolating 414.162.
If you would like Skyline to be able to use those spectra for that peptide, you would need to go to:
Settings > Transition Settings > Instrument
and change "Method match tolerance m/z" to a higher number. (It's currently set to the default .055, which is smaller than the difference between those two numbers).
-- Nick |
| |
| xin huang responded: |
2021-01-21 07:18 |
Hi Nick,
Thanks for the answer. Sorry if I have not made my questions clear enough.
I am looking at the 'Targeted acquisition' file that three of my peptides are shown as the scheduled time window, the precursor and their products. However other two only show the precursor and the time window was 0-14min, not as I scheduled. Is this also because that no MS1 matched with my raw data, thus MS2 are not shown? I have already changed the mass tolerance to 0.5 Da, and re-import the raw data.
Best,
Xin |
|
| |
| Nick Shulman responded: |
2021-01-21 07:36 |
The reason that your other peptides have shorter MS1 chromatograms is that the other peptides have MS2 chromatograms, and when Skyline sees that a peptide has both MS2 chromatograms and MS1 chromatograms, Skyline shortens the MS1 chromatogram for that peptide so that it matches the MS2 chromatograms.
There were no MS2 spectra in your raw file that matched the peptides in your screenshot. Because of this, the MS1 chromatograms span the entire length of the run.
The MS2 spectra in your raw file have isolation windows 414.162, 697.28 and 713.342.
Are you expecting there to be some spectra in there that match the peptides in your screenshot (408.7323 and 529.7851)? I do not see any spectra like that when I look at your file using ProteoWizard SeeMS.exe.
-- Nick |
| |
| xin huang responded: |
2021-01-21 09:01 |
Yes, I am expecting to see spectra of all five peptides. I have made a mixture of five synthetic peptides, and trying to establish MRM methods to detect them at the same time. At least I am seeing the same from Masslynx and Skyline now, I could see MS2 of 414.162, 697.28 and 713.342, three peptides (in Function 2 and 3 in Masslynx), but not the MS2 of 408.7323 and 529.7851 (same here in Skyline). In the beginning I thought there were not enough 'points per peak' when there are overlapping time windows. Then I divided into two injections, once three peptides MRM and another time two peptides MRM, then I could see them all. So there was nothing wrong with the sample, or the MRM method, but somehow not all the spectra are collected? into the Functions?
Anyway, thanks a lot, and I will need to learn how to use ProteoWizard SeeMS as well.
Regards,
Xin |
|
| |
|
|
 Peptide in Function channel 3 from Skyline.png
Peptide in Function channel 3 from Skyline.png TargetedAcquisitionChromatogram.png
TargetedAcquisitionChromatogram.png Peptide VLPQQQAQF targeted acquisition.png
Peptide VLPQQQAQF targeted acquisition.png Peptide GIDTRVGV targeted acquisition.png
Peptide GIDTRVGV targeted acquisition.png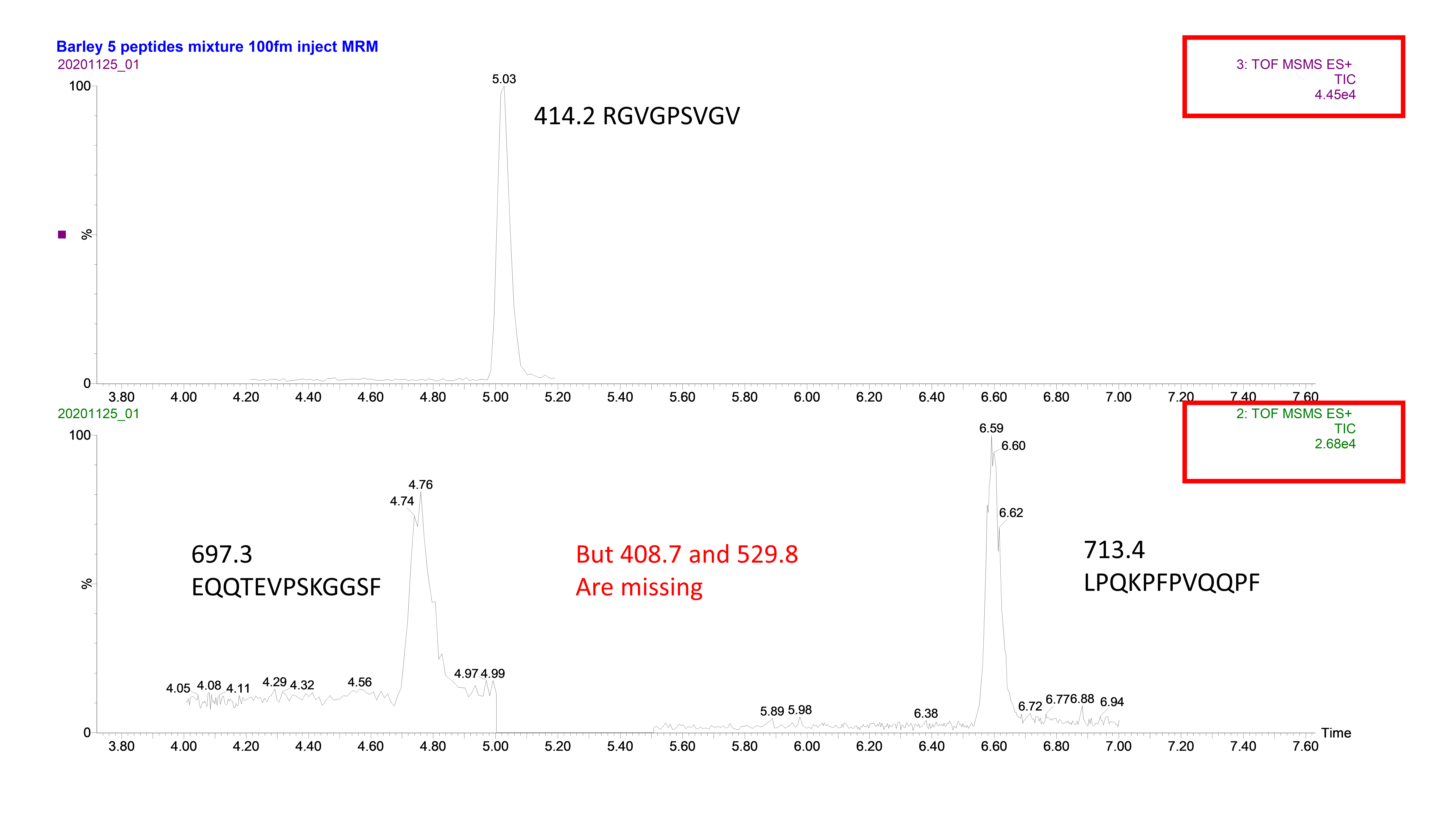 specta.png
specta.png