| Nick Shulman responded: |
2020-12-16 19:18 |
The grey dotted lines indicate that this is a candidate peak which Skyline found, but is not the chosen peak.
One reason that it might not be integrated is that you explicitly told Skyline to remove the peak.
You would be able to see that this is the case by looking at the "User Set Peak" column in the Results Grid when you have a Transition selected.
If you have manually changed peaks, then "User Set Peak" will say "TRUE".
Another reason it might not be integrated is that you went:
Refine > Reintegrate
and had "Only integrate significant q-values checked".
Other than those two things, I can't really think of a reason that things would look the way that you are seeing them.
If you would like, you can send us your Skyline document and I might be able to tell what is going on.
In Skyline, you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file is less than 50MB you can attach it to this support request.
Otherwise, you can upload it here:
https://skyline.ms/files.url
It also might be helpful to see your raw file.
-- Nick |
| |
| henrik molina responded: |
2020-12-17 07:40 |
Hi Nick,
Thx for quick reply.
1) Checked 'user set peak'. All 'False'.
2) Did not 'Refine > Reintegrate'.
3) sky.zip is too large. Wil upload together with two .RAW files (examples of works and not works).
Cheers, h |
| |
| henrik molina responded: |
2020-12-17 07:49 |
Btw: the molecule in question is 'Citrate' ([NEG]>citrate_C0) in negative mode. The experiment is polarity switch. |
| |
| Nick Shulman responded: |
2020-12-17 08:57 |
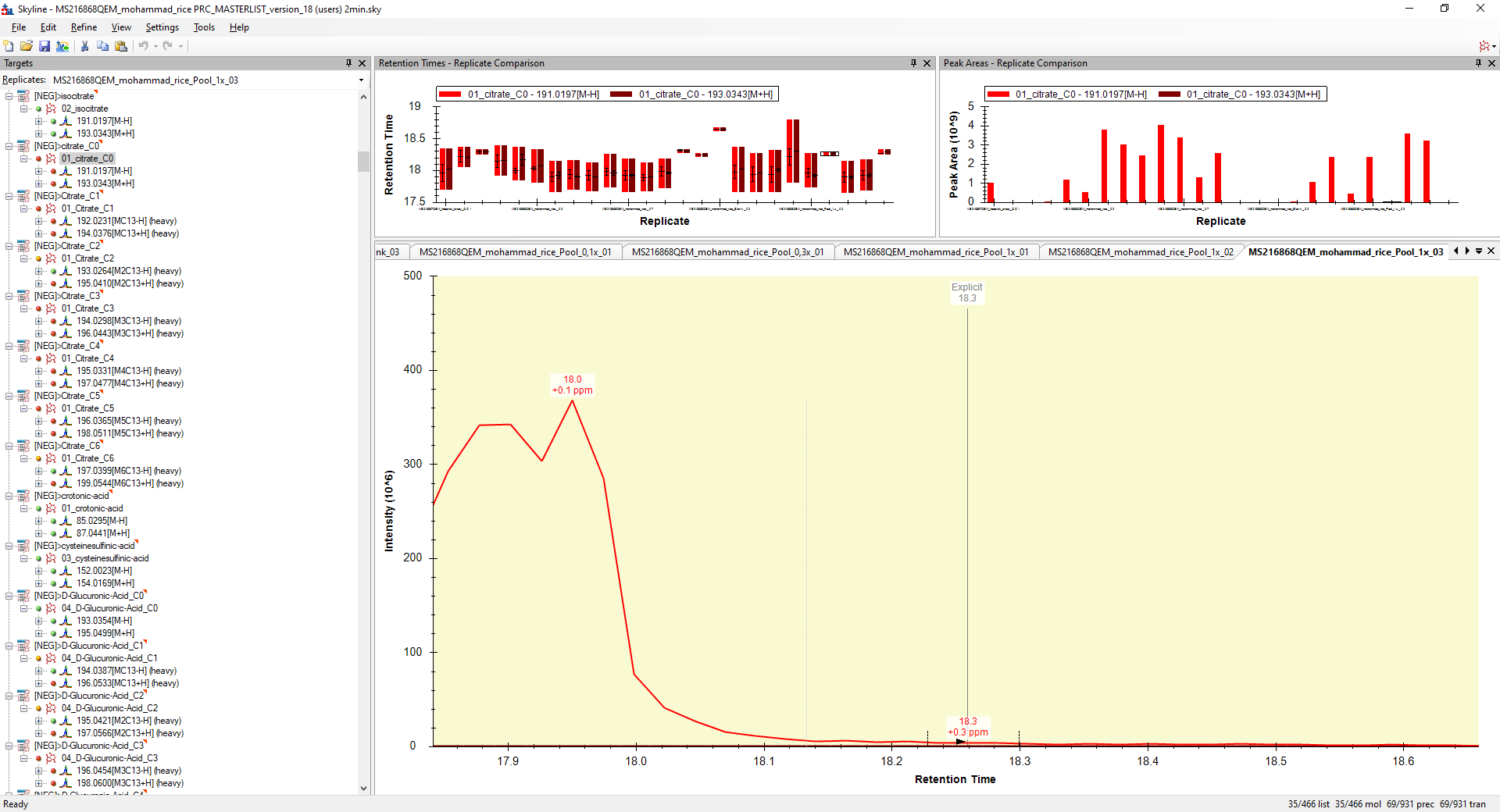
Which replicate were you looking at in that first screenshot? I am not sure I see a chromatogram that looks exactly like what you have.
If you right-click on the chromatogram and choose "Auto-zoom X-axis > Best Peak", Skyline will zoom in on whatever peak it has chosen.
One thing that is a little weird is that the chosen peak in Pool_1x_03 does not look like a peak at all (see attached screenshot "Henrik_Molina_Peak.png")
I believe the reason that this is happening is because Skyline is looking at more chromatograms than you see in the Targets tree.
You can see those extra chromatograms if you right-click on your precursors and choose "Pick Children" and add the M-1, M+1 and M+2 precursor transitions.
However, even after I add those transitions (see attached "Henrik_Molina_withextrachromatograms.png", I still don't see why Skyline would have chosen that spot to integrate. It must have something to do with that being exactly where the Explicit Retention Time is.
The thing that makes me believe that it's these extra chromatograms that are causing the issue is that if I:
1. Go to Edit > Manage Results > Minimize
and check "Discard unused chromatograms"
it will get rid of all of the chromatograms that do not correspond to something in the Targets tree.
And then if I:
2. Go to Edit > Manage Results > Rescore
Skyline ends up picking a more reasonable peak.
It certainly seems like there is a bug going on here which we should investigate. Please let me know which replicate you were looking at in the original screenshot. Also, I would have expected a triangle to be displayed indicating the chosen peak, even if the chosen peak was very small.
-- Nick |
|
| |
| henrik molina responded: |
2020-12-17 09:49 |
My remote access PC just shot down - so will have to wait to access Skyline.
However, can you zoom out so you include 17.5min?
I have attached screenful of m/z 191.01-something.
For my explicit RT I am using a 2min window.
h |
|
| |
| henrik molina responded: |
2020-12-17 13:27 |
Have access to the Skyline file again.
1) 'Discarded unused chromatograms' WAS checked on.
2) Still, I tried to use the re-score function. No difference. Citrate in POOL 03 is still not picked.
cheers, h |
| |
| Nick Shulman responded: |
2020-12-17 13:56 |
"Discard Unused Chromatograms" is not a setting that you turn on and off.
When you go to:
Edit > Manage Results > Minimize
Skyline is offering to do some things to your .skyd file that will make it smaller.
Nothing actually happens until you press "Minimize in place" or "Minimize and save as".
Can you attach a screenshot of what you are seeing (the whole Skyline window)? I am still not sure that I have been looking at the same replicate as you have been.
-- Nick |
| |
| henrik molina responded: |
2020-12-17 18:55 |
It seems as I can click on/of the 'Discard Unused Chromatograms' - see the box in the attached pic.
I am rather sure that we are looking at the same file - see the pic. Can you look at 18min?
h |
|
| |
| henrik molina responded: |
2020-12-17 19:05 |
I clicked the 'minimize in place' and followed up by by a 'manage Results' -> 're-score' => got the integration to work.
A previous 're-score only' did not make any difference. What is the 'minimize in place' doing? Should that be a default function/workflow to use?
Btw: have re-attached the prev. file bc of wrong file extension.
h |
|
| |
| Nick Shulman responded: |
2020-12-17 19:20 |
There is something wrong with that file "4nick_2nd.jpg". Can you try attaching it again?
I am not sure I understand your question.
The "Discard unused chromatograms" checkbox on the "Edit > Manage Results > Minimize" dialog is useful if:
1. You have deleted a bunch of Targets from your Skyline document and you want to make your .skyd file smaller
2. You want to get rid of the extra isotope chromatograms (M-1, M+1, M+2, etc) that Skyline extracted.
3. You imported results from an SRM raw file and there were a bunch of chromatograms that you never wanted.
-- Nick |
| |
| henrik molina responded: |
2020-12-18 06:19 |
sorry for the hick-ups with the Discard Unused Chrom pic. This screen dump should be ok! |
|
| |
| henrik molina responded: |
2020-12-18 10:26 |
Another example. Heavy Arginine (arginine_13C6,15N4 ) in sample ‘_10’ is not picked correctly though it is marked - with grey dotted lines.
Have attached .pdf with example and also included two .RAW files: #10 (problematic) and #11 (ok) - uploaded to your server.
PS: This observation can be reproduced on a different PC using same method (.sky).
And this brings me to a unrelated question – and potential important for the question: We first tried with a basic .sky on the different PC and that gave very different results than the method that I used (have made).
We have compared settings in 'Peptide Settings...' and 'Transitions Settings...' and can not find any differences. Is there a easy way to compare two .sky files?
Cheers, h |
|
| |
| Nick Shulman responded: |
2020-12-18 11:05 |
henrik,
I can tell you that the reason that Skyline is picking the peak at 18.23 minutes in Pool_1x_03 is because the M+2 chromatogram has a one spectrum with nonzero signal at that time. Furthermore, because that peak happens to be at the same location as the Explicit Retention Time, Skyline treats that peak as "better" than all of the other peaks that it finds in the other chromatograms, and everything gets integrated based on that "best" peak.
I think the big thing that Skyline is doing wrong here is that it treats peaks which overlap the explicit retention time as better than peaks that don't, even if the other peaks are still well within the explicit retention time window.
There are other things that maybe also should be addressed:
1. Our peak detection code should not be detecting a peak which only have only one point with nonzero intensity
2. Skyline should maybe not be looking at "hidden" MS1 isotope chromatograms when doing peak detection.
I will ask around and see if there is anything we can do about this behavior.
I cannot tell you what is going on in your latest .pdf file. "sample #10" was not part of the .sky.zip that you uploaded. My guess is that if you were to right-click on the precursors in the Targets tree and choose "Pick Children" and add all the precursor isotopes you would be able to see why Skyline chose the peak that it did.
-- Nick |
|
| |
| henrik molina responded: |
2020-12-18 11:36 |
I will upload the file with the heavy Arg example.
We are not using the M+1 and M+2 bc many of the metabolites has so low isotope signal (and our samples are often already low in amounts) that we think it cause more problems than benefits.
cheers, h
PS: sorry for being so persistent. |
| |
| Nick Shulman responded: |
2020-12-18 12:58 |
For the Heavy Aginine in Sample10, Skyline is actually doing what we would consider the correct thing.
Skyline gives a lot of weight to the number of transitions that had a peak at a particular retention time. If two chromatograms had a detectable peak, that is "better" than if one chromatogram had a large and beautifully shaped peak, but the other chromatogram had no signal at all.
By the way, the Arginine molecule in your document was specified using only a m/z, and no chemical formula. Because of this, Skyline was unable to do the thing of extracting all of other other precursor isotope chromatograms.
Specifying m/z's instead of chemical formulas is a viable workaround if Skyline's behavior of extracting the chromatograms for the entire isotope envelope is causing bad peak picking behavior.
-- Nick |
|
| |
| henrik molina responded: |
2020-12-18 13:50 |
Hi Nick,
maybe a typo, BUT 'heavy' molecules ARE defined by using '. Example: C'6H14N'4O2. Listed under 'Precursor Ion Formula'. Screen shot of doc grid attached.
Just so I understand: for the first example, 'citrate_0', the problem was a M+2 (that was NOT defined as a target but still was used) and by removing (minimize) not used targets/traces the problem was fixed. Should I just extract w/1 peak ('Transition Settings' - 'PEAKS' =1 (original value was se 5)?
Re. the heavy Arginine example. Should I define traces by m/z OR by atomic composition?
cheers, h |
|
| |
| Nick Shulman responded: |
2020-12-18 14:34 |
Oops. You're right. I was confused.
The real reason that Skyline does not extract the M+1 chromatogram for arginine is that the molecule is so small. Skyline only extracts chromatograms for isotopes that are at least 1% of the total (but Skyline also always extracts the chromatogram for the thing that is 1 Dalton less than the precursor).
So, for Arginine, Skyline extracts only the M and M-1 chromatograms. But, the M-1 chromatogram in this case is completely flat and is not affecting peak picking.
The chromatograms that Skyline extracts for you is not affected by the "Isotope Peaks Included" setting at "Transition Settings > Full Scan". You will always get the chromatograms for every isotope whose predicted abundance is at least 1% of the total.
-- Nick |
| |
| henrik molina responded: |
2020-12-23 11:31 |
Dear Nick,
Just to cap this thread:
1) is the overall conclusion that most of these observations are because of a 'bug'? If so, any tricks to deal with them until addressed?
I see them often enough and will be glad to provide examples. Just let me know.
2) how best to compare settings used in different .sky methods?
Cheers - Happy Holiday and all the best for the New Year.
SkyLine rules!!!
h |
| |
 Henrik_Molina_Peak.png
Henrik_Molina_Peak.png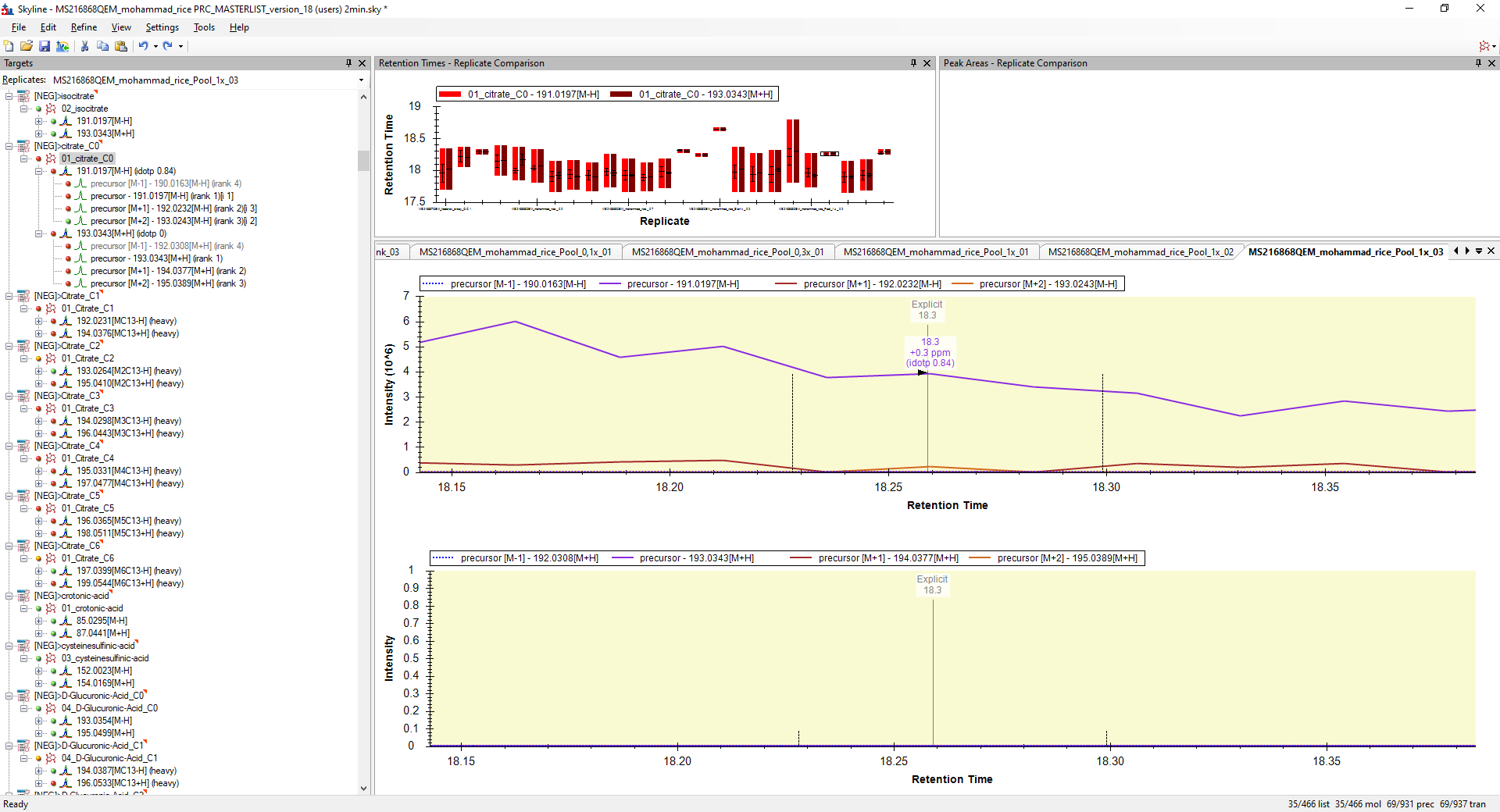 Henrik_Molina_withextrachromatograms.png
Henrik_Molina_withextrachromatograms.png 4Nick citrate negativr mode Capture.PNG
4Nick citrate negativr mode Capture.PNG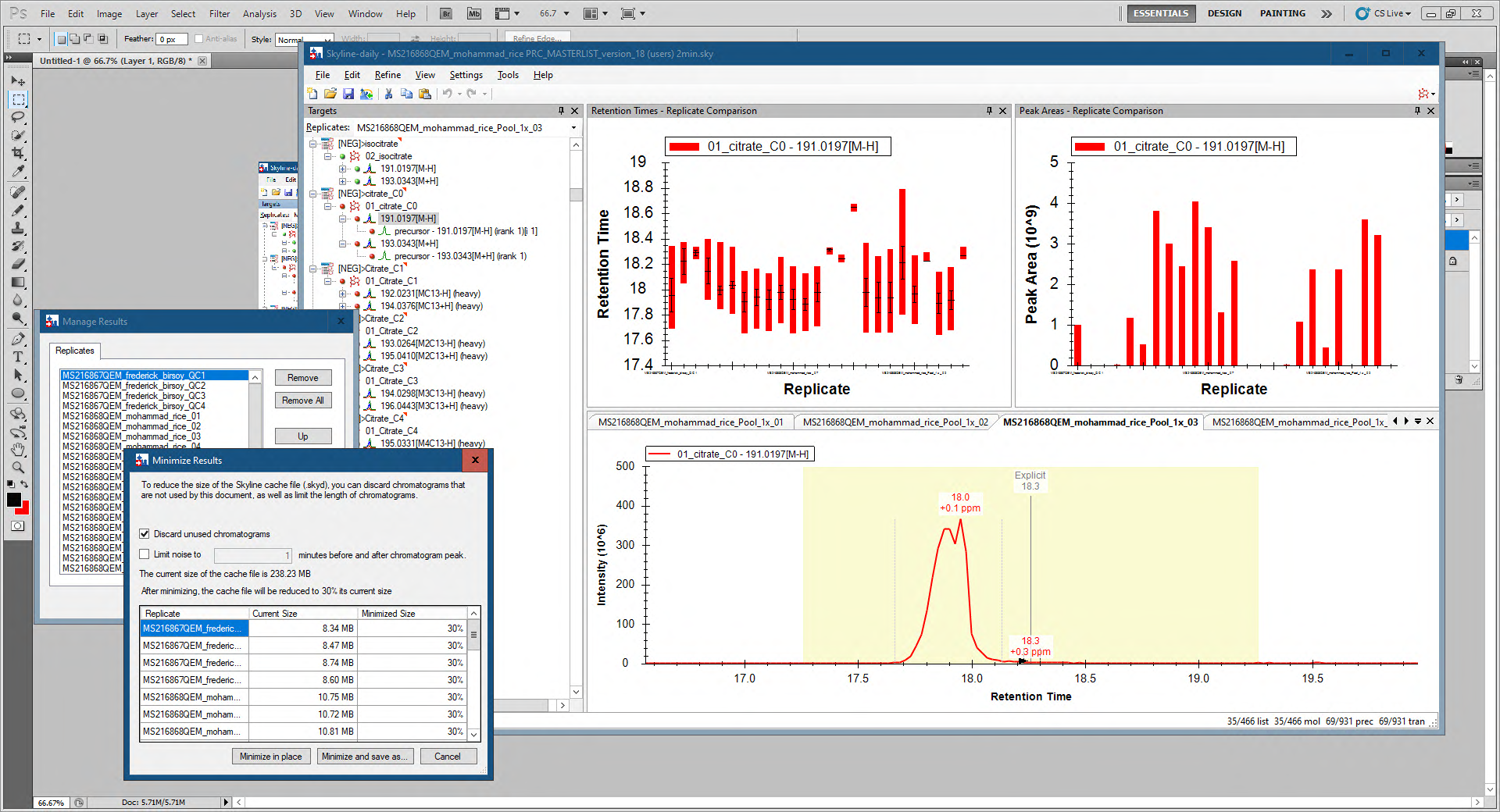 4nick_2nd.jpg
4nick_2nd.jpg after minimize in place followed by rescore.jpg
after minimize in place followed by rescore.jpg.jpg) THIS-ONE (re Discard Unused Chromatograms).jpg
THIS-ONE (re Discard Unused Chromatograms).jpg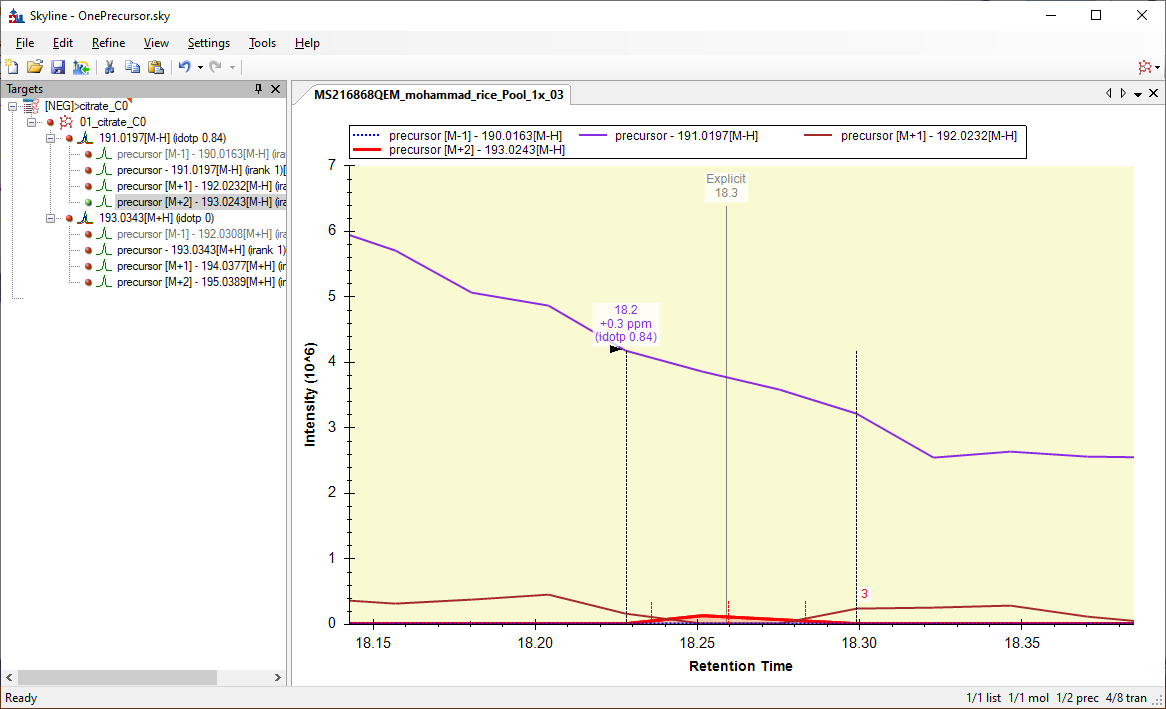 WhyPickPeak.png
WhyPickPeak.png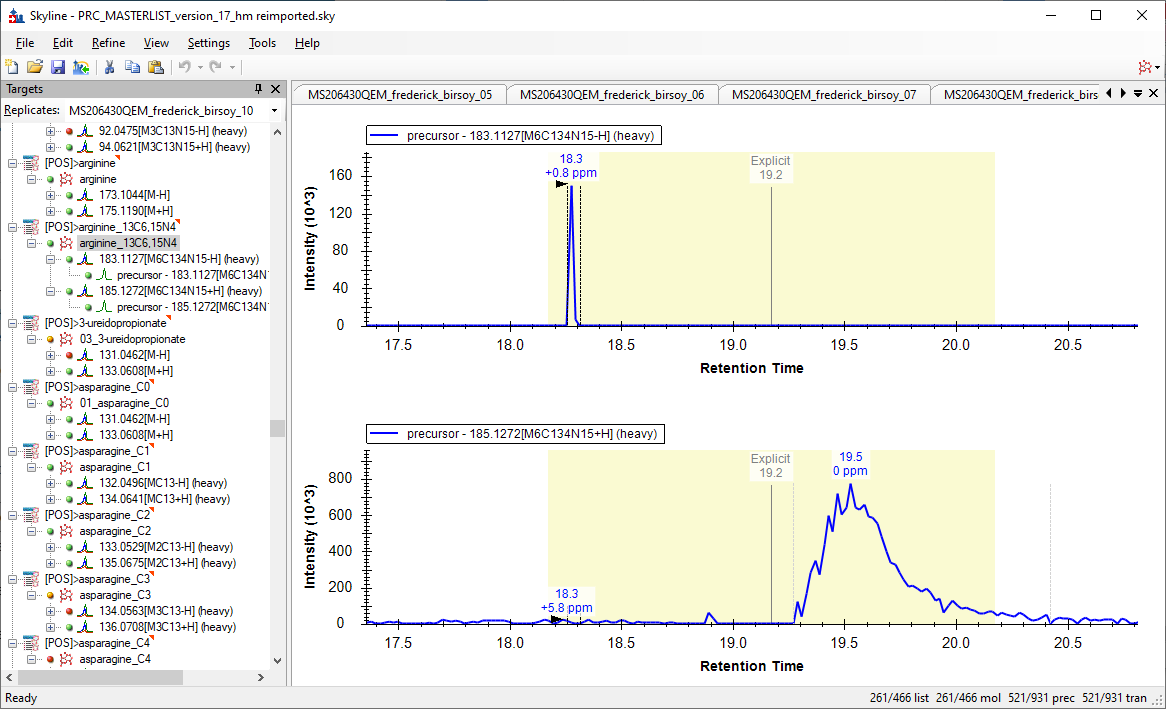 ArgininePeak.png
ArgininePeak.png heavy Arg is defined based on atomic composition .JPG
heavy Arg is defined based on atomic composition .JPG