Ziyang,
No, it is not necessary for there to be MS1 scans.
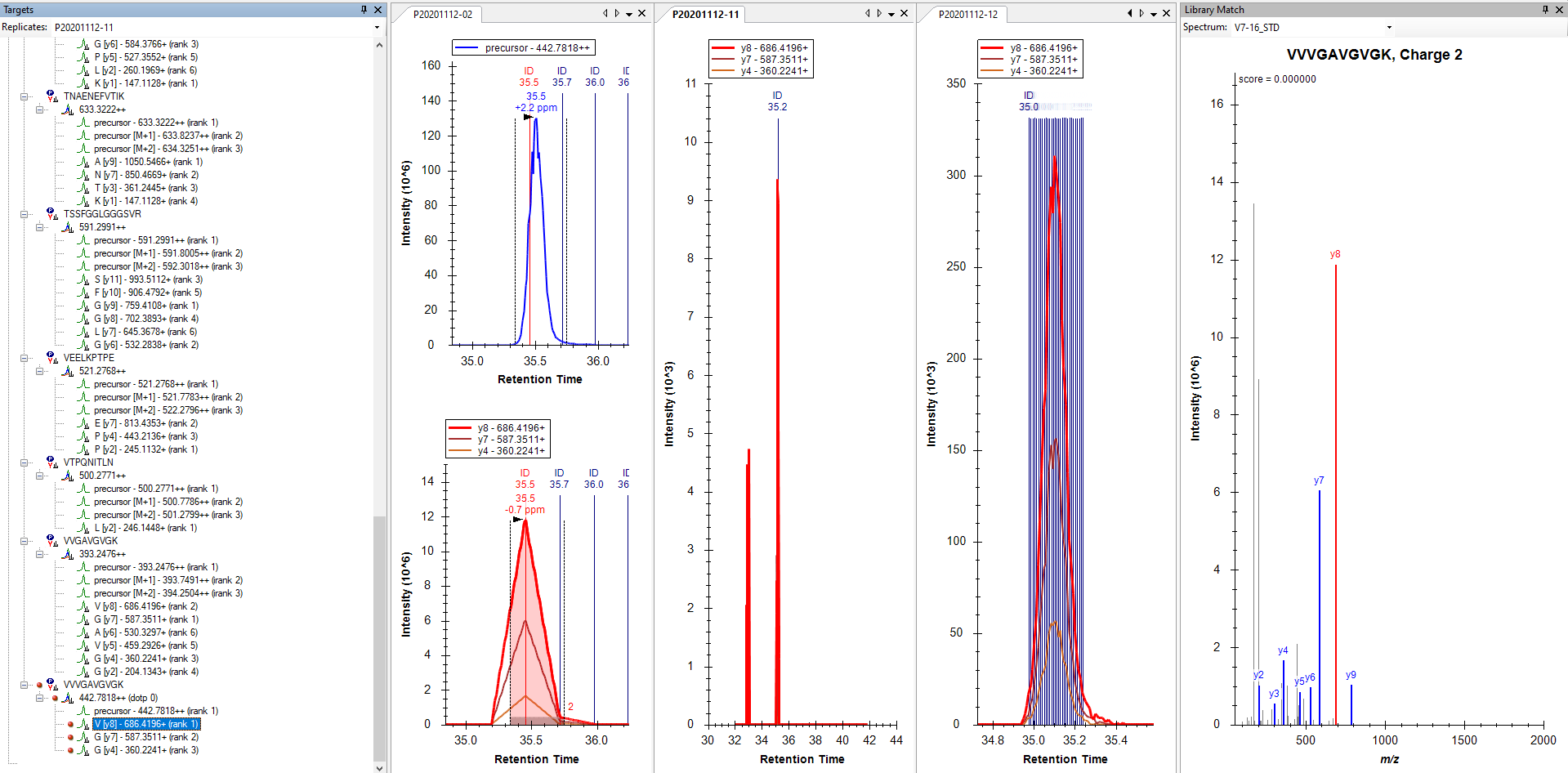
It looks like for most of your precursors, Skyline did not find any matching MS2 scans, and that is why they did not get any chromatograms.
The way that Skyline matches MS2 scans to precursors depends on:
1. The method match tolerance that you have specified at "Settings > Transition Settings > Instrument"
2. The MS/MS filtering Acquisition Method and Isolation Scheme that you have specified at "Settings > Transition Settings > Full Scan"
3. If the Acquisition Method is "Targeted" (i.e. PRM) then MS2 scans will be matched to the one precursor in the document whose mz is closest to what was isolated in the scan. Sometimes this means that one precursor in your Skyline document might steal MS2 scans intended for a different precursor.
I cannot tell which of these things (or maybe something else) is going wrong in your screenshot.
Can you send us your Skyline document and one of your raw files?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file is less than 50MB you can attach it to this support request.
Otherwise, you can upload it here:
https://skyline.ms/files.url
Please also send me one of your raw files that you extracted chromatograms from.
-- Nick
 Screenshot_No_MS1.png
Screenshot_No_MS1.png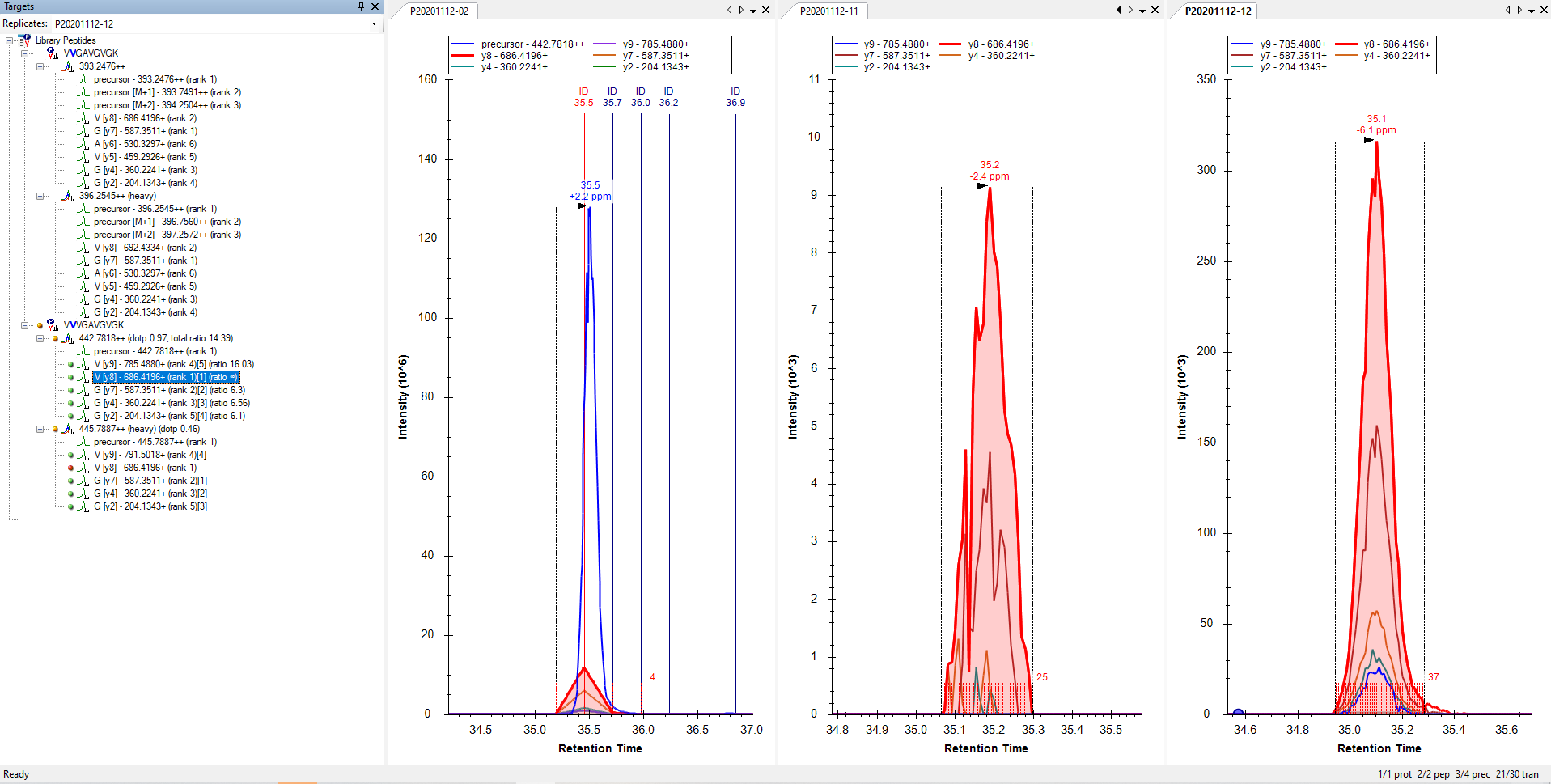 Screenshot_Cleanedup_Lib.png
Screenshot_Cleanedup_Lib.png