| Issue With Retention Time Selection | ed3 | 2020-12-11 06:49 | |||||||||||||||||||||
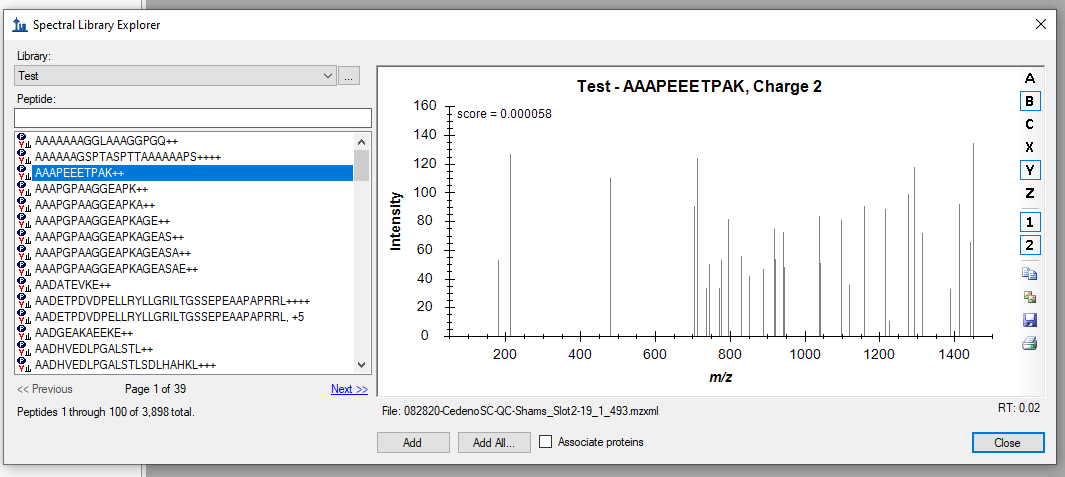
Hello, I am trying to import a pepXML file from PEAKS Online into Skyline to generate a peptide library. However, I am having an issue when doing this as for some reason Skyline is not selecting the appropriate retention time for the peptides in the pepXML file. I have attached a picture of this issue to illustrate what I am talking about. In the attached picture, Skyline has selected the retention time to be 0.02 minutes for the selected peptide; however, the actual retention time of this peptide is 15.20 minutes. Is there a way to resolve this issue? Thank you in advance for your help, and please let me know if any clarification is needed. Best, |
|||||||||||||||||||||||
| |||||||||||||||||||||||
 Example.PNG
Example.PNG