| Incorrect modification (TMT) mass detected in the spectrum file | ab | 2020-11-25 10:25 | |||||||||
Hi, I am trying to build a spectra library from a TMT tagged sample to evaluate and select peptides for a PRM experiment. I defined the peptide and transition settings as recommended in Webinar #17 and included the modification information for TMT at K and N-term. When I explore the built library, I see at he N-term modification is marked as [+228.1511?], the precursor m/z is accordingly 1 Da less than what is reported by my peptide search in proteome discoverer (SEQUEST) and therefore I am not able to evaluate results for any of my proteins/peptides of interest. I am wondering why this is the case. I am including an image as an example. Thank you, Aparna |
|||||||||||
| |||||||||||
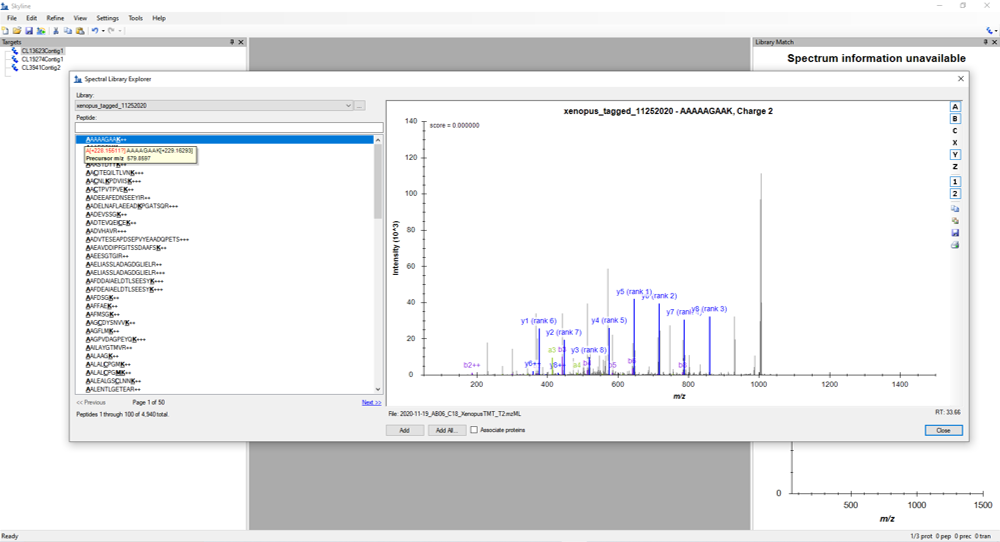 Skyline_TMT_question.png
Skyline_TMT_question.png