| Brian Pratt responded: |
2020-11-09 09:13 |
In earlier versions of Skyline, the Explicit RT value was, as you say, somewhat suggestive. If Skyline didn't find anything that looked like a peak within that window it would go looking elsewhere. Now, though, if it doesn't find anything within that window it just stops looking and records no peak.
It sounds like you wish Skyline would just treat whatever is within that window as signal, regardless of whether it seems peak-like?
Best regards,
Brian Pratt
|
| |
| jrenders responded: |
2020-11-09 09:27 |
Hi Brian,
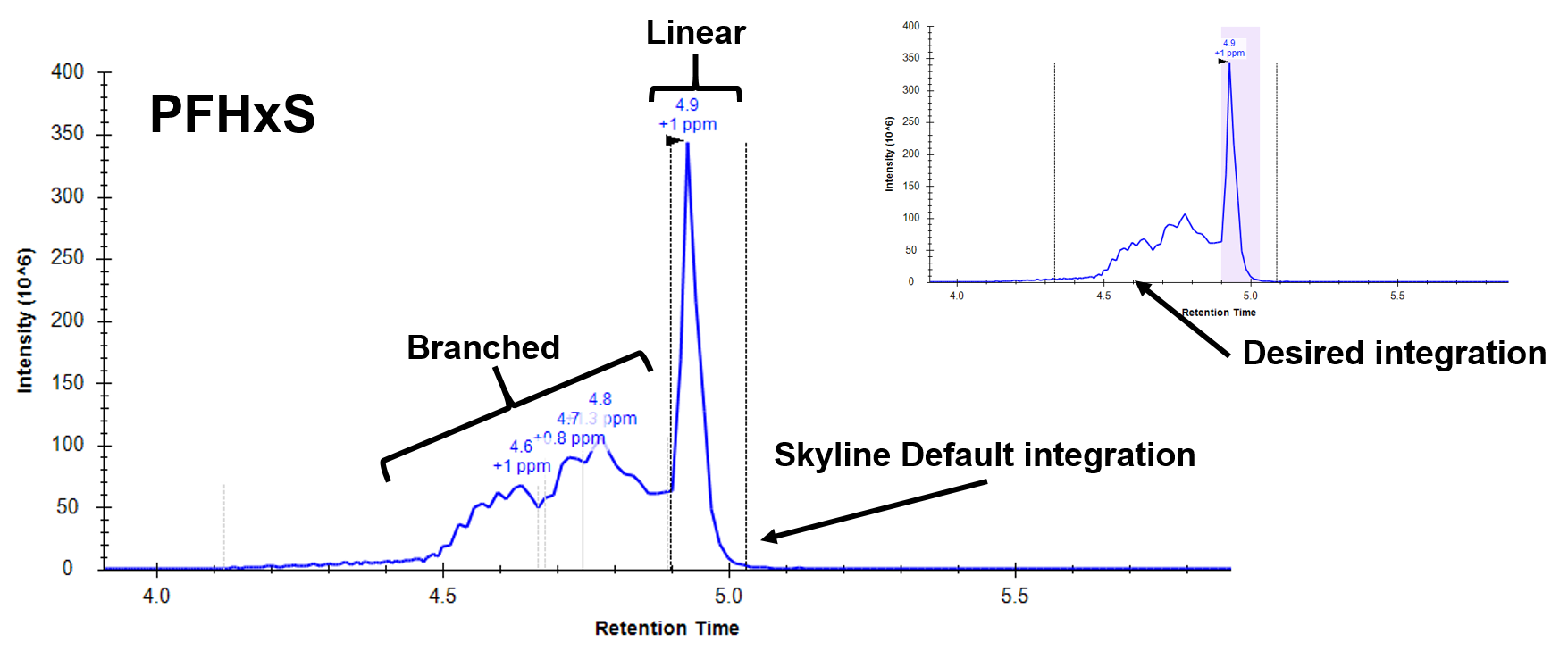
Correct, Skyline's idea of "peak-like" and my idea (in a small molecule world) will likely be very different and will honestly depend on the analyte class. One specific example is in when analyzing PFAS. Some analytes can appear as a branched form or a linear form, with moderate RT differences between the two. Historically, both of these forms are just summed and you report the total. However, some samples will have just linear and some just the branched and some will have both. I imagine this (and other circumstances) would be a hard phenomenon to teach an integration algorithm. If instead, I were able to tell skyline "just integrate everything from 2.3 minutes to 3.4 minutes no matter what" this would save me a TON of time. Then if there is any RT drift or anything else I can go through and manually clean up. Currently, I am going in and correcting every sample (50+) for almost every analyte (30+).
I see some places in the document grid that list what the integration boundaries are but these are return fields and cannot be edited...
Thanks!
|
| |
| jrenders responded: |
2020-11-13 09:44 |
Hi Brian,
Just wanted to follow up on this. Is it safe to say that this functionality isn't available at this time, but may be someday? Thanks.
|
| |
| Brian Pratt responded: |
2020-11-13 10:10 |
I'll make a formal note of it in our development queue.
There are some barriers to implementation, though: Skyline currently makes a pretty hard assumption about the presence of some kind of peak, for the purpose of background subtraction. In this case, where there's signal from your point of view, absent a peak Skyline would see it as noise and subtract it out, leaving something pretty close to zero.
Luckily I believe there is some unrelated work going on in Skyline that might make this more tractable than it formerly was. Could you furnish some examples of the phenomena you're thinking of? Skyline document (File > Share> Complete), raw data, and a few screen shots that illustrate the problem? This would go a long way toward making this a reality.
Best,
Brian Pratt
|
| |
| jrenders responded: |
2020-11-17 09:17 |
Hi Brian,
Thanks for the info - I'll keep an eye out for updates! I've attached some screenshots and a document showing some poor peak shapes that are not handled by Skyline as well as I would hope. Thanks.
|
|
| |
| Brian Pratt responded: |
2020-11-17 11:11 |
Thanks for those images - this is very interesting.
I would have naively looked at those and said Skyline is doing an excellent job of discerning the peak from some major interference. And I'd have (again naively - I'm a software engineer, not a molecular biologist) felt justified because Skyline's integration of the heavy labeled versions seems to be correct.
It almost seems like the branched and linear forms should be treated as different molecules for analysis purposes since they clearly have distinct elution profiles.
What are your thoughts?
Thanks again,
Brian
|
| |
| jrenders responded: |
2020-11-17 11:46 |
Hi Brian,
I definitely agree with you - in a perfect world we would make up a calibration curve for the branched form for and a separate calibration curve for the linear form and handle them separately. The problem, however, is the prevalence of these various forms in unknown samples and the availability of standards. I've observed this phenomenon in two different contexts:
-One is in human metabolism where the body can metabolize a chemical by adding a hydrophilic functional group in one of multiple locations on the [drug] molecule (see attached Image 1). The two molecules that result from metabolism of morphine are isobars and although the mass spectrometer cannot tell the difference (except for sometimes with the fragments if you're lucky), these two molecules will have slightly different retention times. Years ago when we were buying the standard for this metabolite (for running a calibration curve) it ended up that it was only possible to synthesize (or isolate) one of these two variants. Therefore we made a calibration curve with only the one form, but the patient samples contained both forms. In this case we integrated the calibration curve like normal (since there was only a single peak there), but then for the samples we integrated both forms and reported the result as "total" morphine-glucuronide. This is not ideal but is generally accepted for structural isomers (with similar retention times) when an exact standard of each form cannot be acquired.
-In the PFAS world, it is a similar issue, the byproducts that are found in water streams contain both several different branched forms as well as a linear form (Image 2). The standard that we are able to purchase is also a mix of branched and linear because they are unable to sufficiently (or economically) separate the linear form from the various branched forms that may be occurring. The current protocol according to government standards is to integrate the whole mess and label it as a "total" quant that incorporates both forms (so long as ion ratios pass). In the end the toxicology to determine whether the branched or linear forms have a greater impact on human health falls to somebody smarter than I - hopefully they are smart enough to figure out how to synthesize pure standards...
Hopefully this sheds some light on the issues - small molecules can be quite tricky like this at times and that's where the ability to just specify a start and end point to the integration window would be really helpful for reducing manual batch processing.
|
|
| |
| Brian Pratt responded: |
2020-11-17 12:54 |
OK, this improves my understanding of what you're up against.
You might want to experiment with a different document structure, e.g.
PFHxSP
|- PFHxSP_linear
| |- [M-H]
| | [M6C13-H]
|- PFHxSP_branched
|- [M-H]
|- [M6C13-H]
PFHpS_P
|-PFHpS_P_linear
...etc
and assign a different explicit RT to the linear and branched forms of each molecule. |
| |
| jrenders responded: |
2020-11-18 07:14 |
Hi Brian,
This is a good idea, however, this is not the standard established by folks researching PFAS longer than I have. Additionally, when we buy standards at a certified concentration from a vendor, the concentration is certified based on the total branched+linear and so if quant them separately I can no longer confidently assign a quantitative value to my unknowns. |
| |
| Brian Pratt responded: |
2020-11-18 10:06 |
It's just that you're up against some fundamental assumptions in Skyline about the retention time being an important way to tell one molecule apart from another. From Skyline's point of view you've got two different molecules of identical mass, and the chromatography is doing its job of picking them apart.
But of course you know that they're the same molecule from an analysis point of view, so the question before us is how to take advantage of that higher level knowledge. The easiest way would be to somehow get Skyline to combine their signal at the time of chromatogram extraction.
Could I get one or two of your raw files so I can play around with this a bit?
Thanks
Brian |
| |
| Brendan MacLean responded: |
2020-11-18 10:20 |
I will admit that at one point I did realize that there is no inherent reason that Skyline peak integration needed to be limited to just one time range, but that is how I designed it, and after 11+ years, this is really the first potentially valid case anyone has ever brought up that would require multiple time ranges for the same "target". It would not be an easy change to add multiple ranges now. |
| |
| jrenders responded: |
2020-11-18 10:21 |
Hi Brian,
This makes sense and I wouldn't want to change Skyline's integration abilities as all of these exceptions that exist in the small molecule world would just render an integration algorithm useless I would think. What a lot of instrument vendors do in their software is allow you to essentially overrule automatic integration on a case-by-case basis by entering explicit integration windows (for these exception cases), so that you can then apply this window to all samples for a given analyte.
I would be happy to share some raw data, but can you remind me how to share this in a more private forum? Thanks. |
| |
| Brian Pratt responded: |
2020-11-18 10:52 |
I'll contact you off list. |
| |
| jrenders responded: |
2020-11-20 13:00 |
Hi Brendan,
Sorry, I missed your post previously. To be clear, I am still describing only a single time range. I just want to be able to specify that time range explicitly via a number-input text box and have it applied to all samples within a certain molecule assignment. This time range may encompass several peaks (as defined by Skyline), but the range itself is continuous and not broken up into segments or anything (that sounds way more complicated programming/logic-wise). Thanks. |
| |
| jrenders responded: |
2020-11-20 13:02 |
Hi Brian,
Thanks for working that out in the skyline document showing me how I can have both portions of the peak integrated separately. This is an option for getting both portions of the peak integrated automatically (independently), but this would still require me to export and sum the areas and then calibrate outside of skyline which would make my entire workflow pretty much explode. At this point, it is easiest to use CTRL+down arrow and just go through each sample and manually change the integration for each analyte that has this type of irregular peak shape. It's just a shame as this takes a lot of time to do and it seems like something that should conceivably be able to be stipulated once per batch based on an explicit integration boundary setting that overrules automatic integration parameters. I know other folks here at NC State who use Skyline for metabolomics and other small molecule applications that are interested in a setting like this as well because several of them have been asking me if I know how to set explicit integration boundaries (I've become the super-user in our group). I can have them join in the conversation if you think it would be helpful. |
| |
| Brian Pratt responded: |
2020-11-20 15:22 |
Here's an idea to kick around which uses the existing Explicit Retention Time and Window concept, and (I think) would not require major changes to Skyline's internals:
By default, Skyline continues with the current behavior, which is to use the best peak in the specified RT range. If no peak is found in the range, no peak is recorded.
Now imagine a new optional behavior in which Skyline still uses the best peak in the range, but extends that peak's integration boundary to include any other peaks found in the range whose would-be integration boundaries form a continuum with the best peak's boundaries. It is still the case that if no peak is found in the range, no peak is recorded.
So in this new optional mode, for example:
Skyline find peaks A, B, C, D, and E (in ascending RT order) within a precursor's explicit RT range.
B is the best peak, and thus is the peak of record.
A overlaps B in RT so B's boundaries are extended to encompass those of A.
C overlaps B in RT, and D overlaps C. B's integration boundaries are again extended, to also include those of C and D.
E does not overlap D. E is ignored.
If C did not overlap B, then C, D and E would all be ignored and B's boundaries would just be the union of A and B boundaries..
Does that make sense?
Brian |
| |
| jrenders responded: |
2020-11-24 06:11 |
Hi Brian,
This makes sense and would work in some cases, for example the PFAS examples I've shared where there is a very wide low peak semi-overlapping with a larger more resolved peak. However, I think there will still be problem cases because the integration is still predicated on a skyline definition of what constitutes a peak or a peak boundary. If I have two isomeric peaks that I want to integrate as a single compound (i.e., they are isomers of the same compound) and they are baseline resolved, skyline might say these two peaks do not overlap therefore they don't meet the criteria that you've outlined above.
I realize this is not the norm for proteomics workflows and most non-targeted metabolomics workflows but this functionality is not uncommon when developing targeted small molecule assays. If you are doing proteomics workflows you would definitely not want Skyline to confuse adjacent peaks even when they overlap and I really wouldn't want any changes to be made to skyline in this way because I think it would negatively impact many workflows out there.
Right now when I process my batch and I don't "approve" of Skyline's interpretation of my peaks, I go in and manually drag the dashed integration boundary lines to match what I want Skyline to integrate. After I do this for a single sample I am very satisfied with the result for that sample. The problem is that I have sometimes 10's of analytes and 100's of samples that require this manual intervention. Instead of messing with Skyline's inherent integration process/algorithm, isn't there a way to take the integration boundaries I've imposed on one sample and say "copy these exact integration boundaries to all other samples for this target"? In this way the user would overrule/ignore Skyline's integration and go with the user's defined boundaries. Right now this workflow functions just fine within Skyline, the issue is that it can only be implemented one sample at a time via dragging integration boundary lines. It just seems like a target-level strict RT boundary assignment text box entry or rt-click "apply integration boundaries to all" function should be feasible without impacting the Skyline integration brain.
The folks I work with were certain that the "Explicit Values" --> "Retention time" and "Retention time window" functionality within the "Modify..." command (rt clicking a target in the target window) would do what I am describing but were confused when Skyline still tried to enforce it's integration rules within these explicitly user-defined RT boundaries. |
| |
| Brian Pratt responded: |
2020-11-24 09:00 |
>> I really wouldn't want any changes to be made to skyline in this way because I think it would negatively impact many workflows out there.
Absolutely, any behavior change would need to be something the user has to explicitly enable.
>> isn't there a way to take the integration boundaries I've imposed on one sample and say "copy these exact integration boundaries to all other samples for this target"?
Have you looked at the "Apply to All" function?
>> The folks I work with were certain that the "Explicit Values" --> "Retention time" and "Retention time window" functionality within the "Modify..." command (rt clicking a target in the target window) would do what I am describing
I could imagine a (specifically user enabled!) mode where we don't even worry about overlapping peaks, but take the whole window if any peak at all is found. The problem with taking the whole window whether or not a peak is found, as mentioned previously, is that Skyline will likely get tripped up on background subtraction. I suppose if we find no actual peak we could just declare the "peak" at the maximum intensity RT for the window, and hope for the best. |
| |
| jrenders responded: |
2020-11-30 12:51 |
Hi Brian,
I have been trying unsuccessfully to use the "Apply to all", it appears to also be restricted to Skyline's definition of what constitutes a peak (or tries to find and use certain inflection points) and therefore does not actually apply what I have done to the first sample to all other subsequent samples.
I'm not certain how background subtraction works in these workflows? When I manually specify integration window boundaries (by dragging the dotted lines), does Skyline not just integrate everything above the baseline and use that area as the "peak area"? I wasn't aware of any subtraction taking place in these measurements. Maybe my lack of understand of this subtraction process is what is tripping me up here because I still don't understand why Skyline has to "find a peak" can it not just integrate everything above the two points on the spectrum dictated by my manually manipulated RT boundaries?
Thanks for putting up with my questions =). |
| |
| Brian Pratt responded: |
2020-11-30 15:32 |
OK, it sounds like we won't get to where we need to be without some code changes.
I haven't actually looked into the background subtraction code, but in discussion with the other Skyline devs I was given to understand that there would be issues with the current implementation without a clear sense of a peak. Presumably this could be worked around but as I say I've not gone into the details so I don't know how much effort that might entail, sorry.
Best,
Brian |
| |
| wlstutts responded: |
2021-07-14 13:21 |
Hi Brian,
I too would like to be able to explicitly specify a RT range for peak integration. I work with lipids which have tons of isomers that are often closely eluting. The "explicit RT" functionality currently available in Skyline does not work well for many of my lipidomics datasets with large numbers of isomeric peaks that often have close elution times. Allowing the user to over-ride the peak detection algorithm would be helpful in many cases. Perhaps there could be a checkbox in the "modify molecule" window under "explicit values" that would over-ride the peak detection and simply integrate the peak area over the user-specified RT window.
Thanks,
Whitney |
| |
| Brian Pratt responded: |
2021-07-19 14:28 |
Hi Whitney,
Thanks for the input. As previously noted, an option to force integration over an RT range that might be nothing but noise could be problematic, but we'll take this under consideration.
Best,
Brian |
| |
| wlstutts responded: |
2021-07-19 15:03 |
Thanks Brian! Do you know why the "apply peak to all" often fails to integrate the correct peak (within the specified expected RT) even when there is a peak with an apex at that RT? I often get an error message (attached) that refers to a RT that is not the expected RT and there is often a nicely shaped peak at the expected RT. I am attaching a few files from this dataset; you can see the two chromatographically separated isomers (one file from a standard mixture and one from a representative sample), but for some reason I am not able to used the expected RT setting to correctly integrate these compounds separately. Any advice appreciated! |
|
| |
| Kaipo Tamura responded: |
2021-07-19 18:07 |
Hi Whitney,
The algorithm behind "Apply Peak to All" does sometimes fail. It uses some simple heuristics that were originally designed before Skyline had small molecule support, so the performance is also likely poorer than with peptides.
One thing which may be an issue here is that the area of the peaks are a factor - peaks with <10% of the total chromatogram area are not considered and the percent area can affect which peak is chosen as a match (although much less important than the retention time).
Also, the feature you have requested is under development and should hopefully be available not too long from now.
Thanks,
Kaipo |
| |
| Juan C. Rojas E. responded: |
2021-07-20 01:06 |
Hi all,
What about using the File->Import->Peak Boundaries... option?
This oversimplistic override of Skyline's smart peak detection algorithm would only work if you have really stable chromatography, no close eluting interfering species (besides the multiple forms of your analyte of interest), and (up to some extent) not limited by file size.
1. Import results with "Include all matching scans" and integrate results of a representative sample.
2. Export the default "Peak Boundaries" report.
3. With a text editor, excel, or a programming script, make as many copies of the rows as the number of files you wish to integrate while changing the "File Name" for each "copy". Export the results as a .csv so Skyline can read it back.
4. Import all other files of interest with the "Include all matching scans"
5. Import your "copy-paste" Peak Boundaries by File->Import->Peak Boundaries...
6. Check the results make sense since a lot peak detection overrides are being force with this approach.
I hope this "trick" helps.
Sincerely,
JC
PS: jrenders case woule be great to test with LC-IMS-MS. It is possible those isomers have different CCS. |
| |
| jrenders responded: |
2021-07-20 08:32 |
Hi all,
Thanks for the conversation around this. I'm glad to head there may be some support for forced integration boundaries in the future.
Re: Juan's suggestion: This sounds like a great idea but I hit some snags trying to implement. The default Peak Boundaries report doesn't include any molecule information (just "peptide modified sequence", which is blank for small molecule work). I tried exporting the report in default form (without any molecule information) and with some additional columns to help to read the table. I modified the csv and tried reimporting but I get an error "The following molecule in the peak boundaries file was note recognized: (BLANK) Continue peak boundary import ignoring this molecule? OK/CANCEL". I get this message with and without the additional columns. Hitting okay does not change the integration times shown in the table either way. "Include all matching scans" is already selected on my transition settings...
I can send a .zip if that helps.
Regarding the CCS measurements see: Anal. Chem. 2020, 92, 6, 4427–4435. I am working with Erin Baker on the data in question here.
Thanks. |
| |
| Juan C. Rojas E. responded: |
2021-07-21 02:14 |
Hi jrenders,
I had not realized that the default "Peak Boundaries" report was tailored specifically for proteomics work only as of late I do proteomics only so I hadn't noticed this caveat...
(Disclaimer: I am just a curious user so a lot of what I say is speculation of how I have come to understand how Skyline works)
The peptide modified sequence is used to determine the precursor ion molecular formula using the amino acid sequences and the PTMs stored in the skyline document (for modified peptides). Then uses the specified RT times to report the integration boundaries for all the ions associated to the monoisotopic mass of that precursor. I tried to create a custom report by replacing the Peptide Modified Sequence with the "Molecule", "Precursor Ion Formula", or "Precursor" and replace the column name from the reported .csv so Skyline would allow reading it back...however in all cases it failed because Skyline is really expecting to read a peptide sequence.
I am sorry I can't help more here since it depends on the programming of the software. However, as Kaipo pointed out, this might all be resolved soon with the functionality pointed out by Whitney.
Erin Baker's work and how I think you want to use IMS here is absolutely brilliant! If the CCS of your mutiple conformers is quite different then by applying an IMS filter to the integration would override a lot of these issues you are facing currently. However, I do wonder if Skyline will like/allow having multiple isomers with different drift time integration windows. I had some issues with isomers before:
https://skyline.ms/announcements/home/support/thread.view?entityId=74319f52-411b-1039-ba2c-e465a3931f5b&_docid=thread%3A74319f52-411b-1039-ba2c-e465a3931f5b
The seminar Erin and Brendan gave have allowed me to implement LC-IMS-MS/MS on a daily basis for my work. If you ever feel discussing issues about these topics can help I would be happy to share my experiences so e-mail me if it comes to that: juan_camilo.rojas_echeverri@uni-leipzig.de
Sincerely,
JC |
| |
| jrenders responded: |
2021-07-22 08:23 |
Thanks for your insight Juan! This is good info. |
| |
| wlstutts responded: |
2021-08-04 10:02 |
Hi All-
Glad to hear that this new functionality is under development! This will be extremely beneficial to many of us on the small molecule side.
Thanks,
Whitney |
| |
| viragsagikiss responded: |
2021-10-07 08:04 |
Hi all,
Just wanted to follow up on this as we have contacted about peak boundaries before.
Our group have run into the exact some problem as described above by jrenders, and thanks for describing it so well (although the reason is different, smeared peaks and highly matrix specific peak shape, changing peaks shapes during the batch with ion-pairing chromatography)
" Right now when I process my batch and I don't "approve" of Skyline's interpretation of my peaks, I go in and manually drag the dashed integration boundary lines to match what I want Skyline to integrate. After I do this for a single sample I am very satisfied with the result for that sample. The problem is that I have sometimes 10's of analytes and 100's of samples that require this manual intervention. Instead of messing with Skyline's inherent integration process/algorithm, isn't there a way to take the integration boundaries I've imposed on one sample and say "copy these exact integration boundaries to all other samples for this target"? In this way the user would overrule/ignore Skyline's integration and go with the user's defined boundaries."
To solve it, we constantly using peak boundaries to overcome this, so it is possible in the Small Molecule world, and yes it often just integrate noise, but that's also something we need for the statistical analyses instead of zeros.
Feel free to contact me if you need more details on how this is applied.
Best regards,
Virag |
| |
| Kaipo Tamura responded: |
2021-10-07 16:05 |
Hi Virag (and everyone),
The next Skyline-daily release will contain a feature called "Synchronize Integration", where changing the integration on one replicate will copy those changes to another selected set of replicates.
Hopefully, it should help with some of the issues discussed in this request.
Thanks,
Kaipo |
| |
| Juan C. Rojas E. responded: |
2021-10-08 01:10 |
Life just became a bit sweeter with these "Synchronize Integration" news! Can't wait to test it!
Thank you!
JC |
| |
| viragsagikiss responded: |
2021-10-08 02:10 |
Wonderful news! Very excited to see, thank you, Skyline already makes my work so much easier and this feature could be game changing in our work-flow.
Virag |
| |
| jrenders responded: |
2021-10-08 07:02 |
Kaipo,
That is excellent news! Thank you so much for sharing that info!!! Manually synchronizing integration takes me hours on large sets. This is great! |
| |
| wlstutts responded: |
2021-10-08 07:31 |
Great news! Thanks so much to everyone who helped make this happen! |
| |
| viragsagikiss responded: |
2021-11-03 10:54 |
Hi all,
Is it the best post to give feedback on this?
I don't know, but i just want to confirm that synchronised integration is everything we had hoped for.
It does exactly what we use: apply certain peak boundaries for all samples.
Just much easier and faster with one click, instead of us crafting together a peak boundaries CSV with all Molecule and File names.
Thank you!!!
Virag |
| |
| Kaipo Tamura responded: |
2021-11-03 11:48 |
Hi Virag,
Thank you for the feedback, nice to hear that it is working well for you. We are still working on some bugs and improvements on it.
Thanks,
Kaipo |
| |
 Skyline_Small_Molecule_Integration.png
Skyline_Small_Molecule_Integration.png Skyline_Small_Molecule_Integration_02.png
Skyline_Small_Molecule_Integration_02.png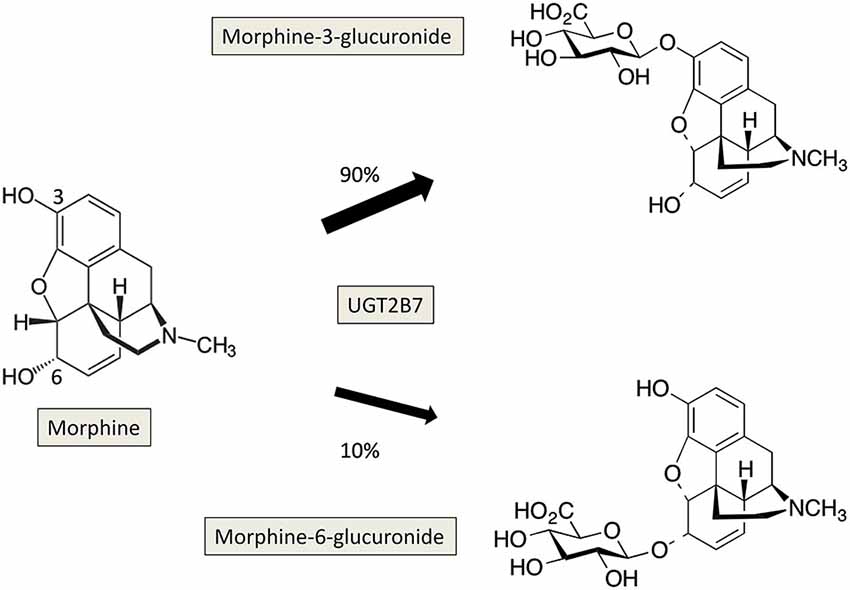 Image 1.jpg
Image 1.jpg Image 2.jpg
Image 2.jpg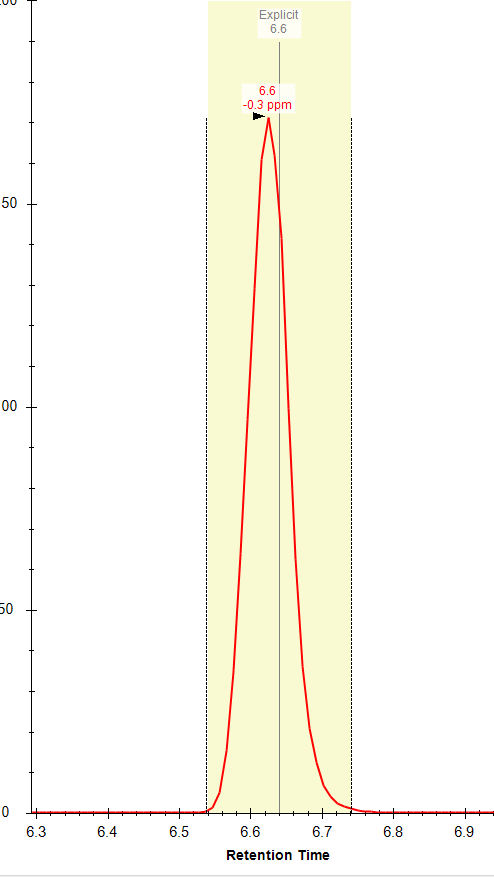 Capture2.PNG
Capture2.PNG Capture.PNG
Capture.PNG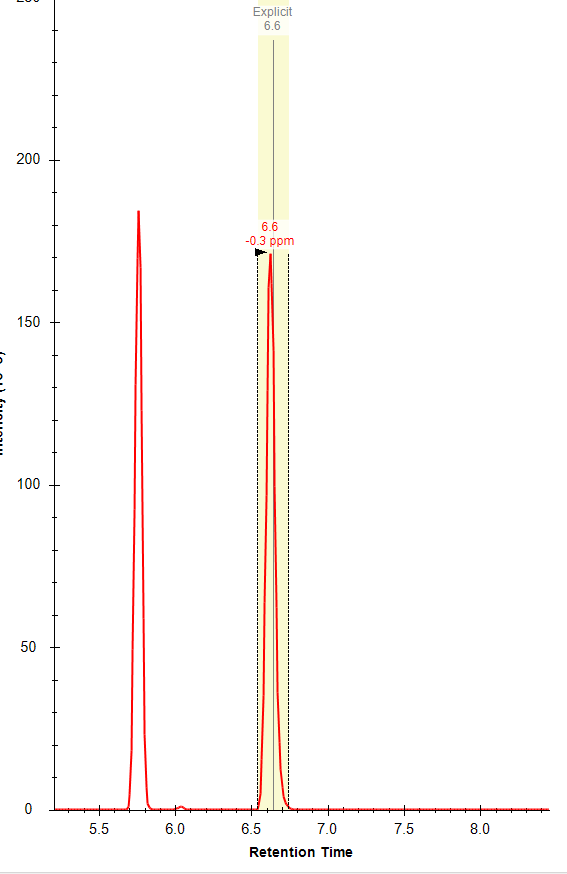 Capture3.PNG
Capture3.PNG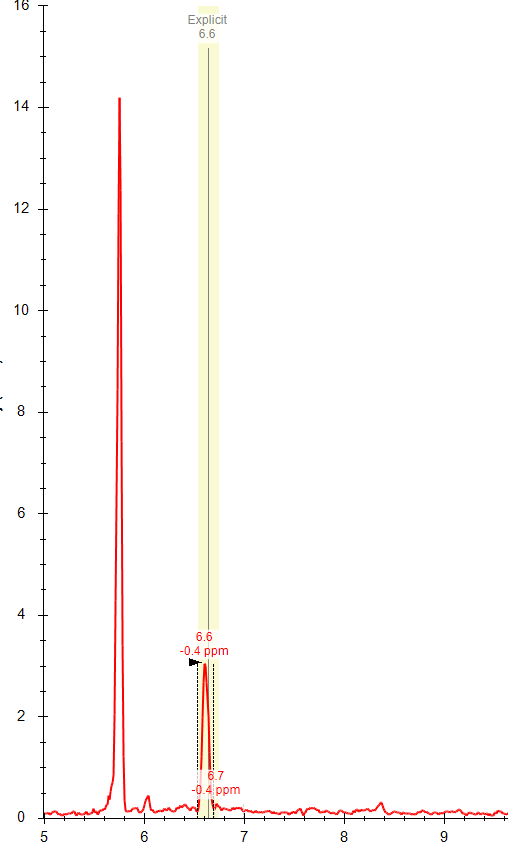 Capture4.PNG
Capture4.PNG