Hi Joerg,
I'm not seeing this on the Fiehn lab website. Perhaps you meant MoNA? The spectrum counts agree, so I'll assume that's the case:
There are a couple of issues with this file, which I would be very pleased to work through with you.
The first issue is that many spectra (6500, actually) provide no hint as to precursor adduct e.g. 1-Methylhistidine at line 520.
When Skyline encounters incomplete entries like this, it just skips over them. I'll add warning messages about that, but it would be better if we could actually use these entries instead of discarding them. Is there an assumption we can safely make instead of dropping these? If we can assume z=1 from "Ion_mode: P", should we then assume protonation (m/z = mass + H), or inherently charged (m/z = mass)?
The other issue in this file is that there are ambiguous entries, as Skyline warns:
Imported library contains multiple entries for one or more molecule+adduct pairs.
This is probably due to the library containing entries for multiple parameters such as instrument type or collision energy, but Skyline keys only on molecule+adduct so these entries are ambiguous.
You should filter the library as needed before importing to Skyline
The important point there is "You should filter the library as needed". For example, Glycerol has four entries, three of which are "Instrument_type: Quattro_QQQ" with various CE values, and the fourth is "Instrument_type: GC-MS". For better or worse, Skyline's spectral libraries don't track instrument type or CE so you wind up with four library entries that are identical except for their fragments. That's probably not what you want.
We originally dealt only with NIST .msp, and NIST does come with convenient filtering tools. But if we're going to accept these kind of random database dumps then we should think about how Skyline could help with the filtering at import time. I'd be happy to hear any ideas you might have on that.
Best,
Brian
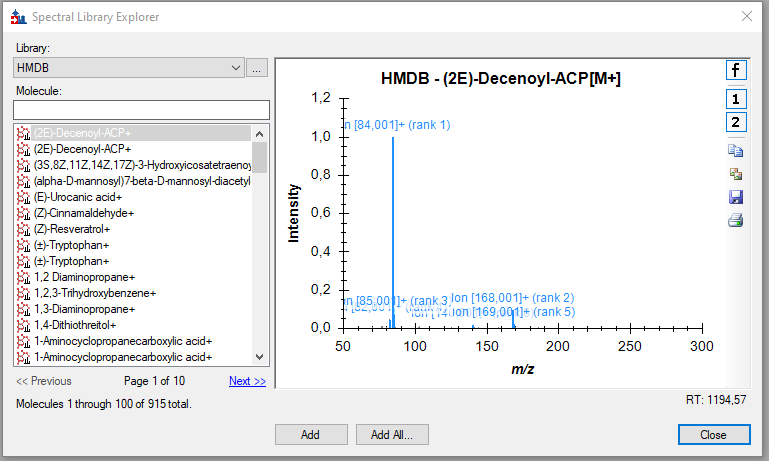 HMDB_import1.png
HMDB_import1.png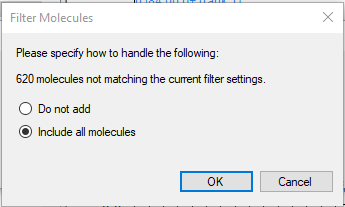 HMDB_import2.png
HMDB_import2.png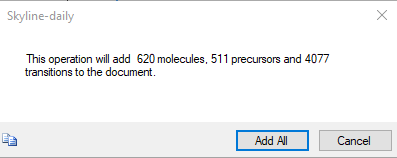 HMDB_import3.png
HMDB_import3.png HMDB_import4.png
HMDB_import4.png