Hi,
We imported 4 .raw files into skyline, and used a bibliospec file to import all the sequences into skyline.
We then created an inclusion list for all the peptides of interest (~700) for PRM.
However, the RT in the inclusion list are all off if I select "average RT", since the peptides selected were not found in all datasets. The other option is to use a single dataset out of 4, but then the RT will be off for the peptides that were not found in that specific dataset.
The best way would be to use the RT from the dataset that matches the spectral library. Is there a way to do this, am I missing something? If not, is it possible to add this as an option in future release?
This would be great, as of right now, we are adjusting the RT window manually for each peptides in each datasets...
OJ
|
| |
| Nick Shulman responded: |
2020-09-17 10:36 |
Can you send us your Skyline document?
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms and spectral libraries.
If that .zip file is less than 50MB you can attach it to this support request. Otherwise, you can upload it here:
https://skyline.ms/files.url
I am not very familiar with all of the ways that it is possible for Skyline to predict the retention times when exporting methods, transition lists, or isolation lists. Where are you seeing the option to use "average RT"?
Did you build your BiblioSpec spectral library from peptide search results? If you want to use the retention times in the .blib file for scheduling, it might have been necessary to create an iRT predictor at the same time that you are building the spectral library. (Skyline-Daily has a very easy choice "Automatic" for 'iRT Standard Peptides" on when building a spectral library)
If you had an iRT predictor built from your spectral library, you would want to make sure that "Use measured retention times when present" is unchecked at "Settings > Peptide Settings > Prediction". Also, you might need to manually specify a particular slope and intercept at:
Settings > Peptide Settings > Retention time predictor dropdown > <Edit Current...>
-- Nick |
| |
| ojulien responded: |
2020-09-17 13:17 |
Hi Nick,
I really want to use the observed RT ("Use measured retention times when present"), not a predicted one.
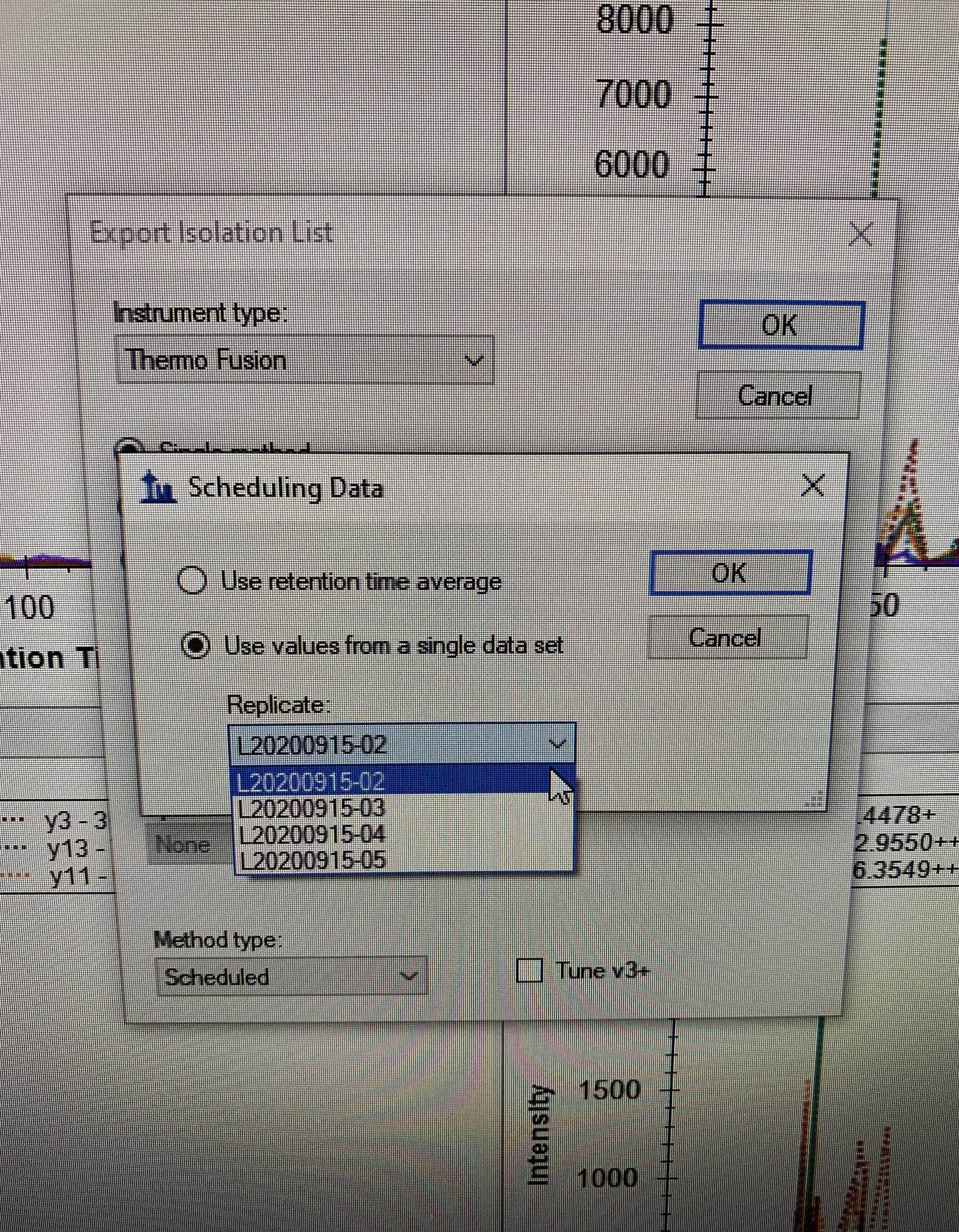
I can select average vs single RT when I export the inclusion list (see attached snapshot).
I also attached a snapshot showing the library window, showing the spectrum/peptide from a given dataset. So skyline "knows" which dataset to use already for each peptide, I just don't have the option to select it at the end
So ideally, we could have a third option when exporting the inclusion list (snapshot #1), which would be something like "Use value from Library". LEt me know what you think. Happy to share the skyline files, but they are 2.5 GB... let me know.
OJ
PS. Yes, I exported my .blib file for a search containing RT, etc. |
|
| |
| Nick Shulman responded: |
2020-09-17 13:41 |
It sounds like what you want to do is remove the peaks from your Skyline document where the peptide was not detected in a particular replicate. In that way, when you take the average of the retention times where the peptide was detected in your .blib file. (Actually, almost the same. The retention times of the peaks in your Skyline document are the apexes of the chromatogram peaks, whereas the retention time of a peptide spectrum match is usually closer to the beginning of the peak).
You can use the Document Grid to remove all of the peaks matching a certain criteria. To do this, view the Document Grid:
View > Document Grid
Then, create a report with the columns "Peptide", "Replicate", and "Identified". ("Identified" is under "Proteins > Peptides > Precursors > Precursor Results")
Then, filter the document grid so that it is only showing you rows where "Identified does not equal TRUE".
Select all the rows in the Document Grid, and then use the Actions dropdown at the top of the document grid to choose "Remove Peptide Peaks".
This will remove all of the peaks where the peptide was not found in a particular replicate.
It sounds like you might be wanting to use the exact time where the peptide was found in the spectrum that is displayed in the Library Match window. The Library Match window shows you the highest scoring spectrum from your peptide search results. I cannot think of a way to end up with only the peaks left where they came from the highest scoring peptide search match, but I am also not sure why you would want to do that.
Certainly, it is no trouble if you want to send us your 2.5GB Skyline document.
-- Nick |
| |
| ojulien responded: |
2020-09-17 17:54 |
That might work. We will give it a try and report back next week. Thanks a lot for your help. |
| |
| Brendan MacLean responded: |
2020-09-22 16:02 |
I agree that what Nick describes might be the closest to what you have suggested that you could currently get from Skyline, and I have considered having an option like this for extracting retention times into iRT libraries, that is to only use the measured RTs for peaks with MS/MS IDs in them. My motivation was working with factionated data where not only are peptides not identified in all runs, they are not expected to be present in all runs. Otherwise, not necessarily being identified in all runs does not mean that Skyline cannot find a perfectly good chromatogram peak through its retention time alignment.
Also, you should prefer to use the chromatogram peak times over the MS/MS spectrum times, which would be less representative of the chromatogram peak apex you are aiming to capture with your scheduling window.
But, as Nick suggests, you can use the Document Grid to bulk remove integration for peaks and that should remove them from the "average RT" which Skyline uses.
Thanks for your posts and screenshots.
--Brendan |
| |
| ojulien responded: |
2020-09-23 11:48 |
Hi Brendan and Nick,
Thanks for your time. So we tried to filter the Document as suggested above, and it works for half of the peptides; so that's good. Unfortunately, it removed the RT for ~half of our peptides. It is still faster than having to adjust the RT for all our samples. All we have to do now is to re-add the desired ID back on our list, and then we will export our list and everything should be fine.
Having the option discussed above would still be the best way to go I think (i.e. only use the measured RTs for peaks with MS/MS IDs in them.), especially when dealing with fractionated data as Brendan mentioned.
For now, we can move forward with this project, so I thank you both for your help. Let me know if you ever add this option later on. :)
Best,
Olivier |
| |
|
|
 Untitled 2020-09-17 at 2.08.08 PM.jpg
Untitled 2020-09-17 at 2.08.08 PM.jpg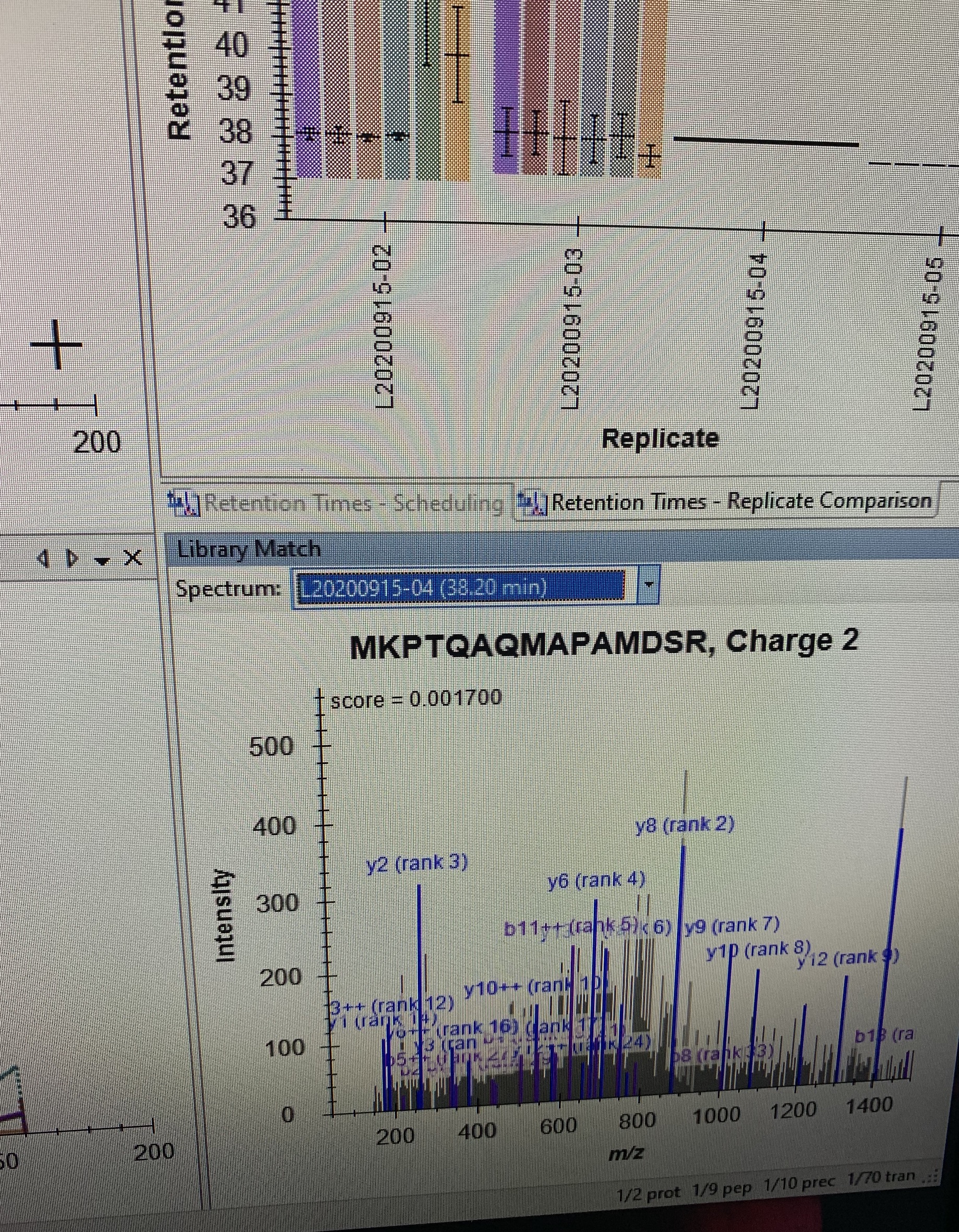 Untitled 2020-09-17 at 2.08.13 PM.jpg
Untitled 2020-09-17 at 2.08.13 PM.jpg