Hi There,
I am running a small molecule calibration curve using skyline. On my instrument, I am acquiring data via an inclusion list, so MS1 data matching to my list triggers an MS2 event for that mass. This inclusion list is also scheduled but my RT's are set correctly with 1.5 min of buffer on each side of the peak (on a 10 min method - so a pretty wide window).
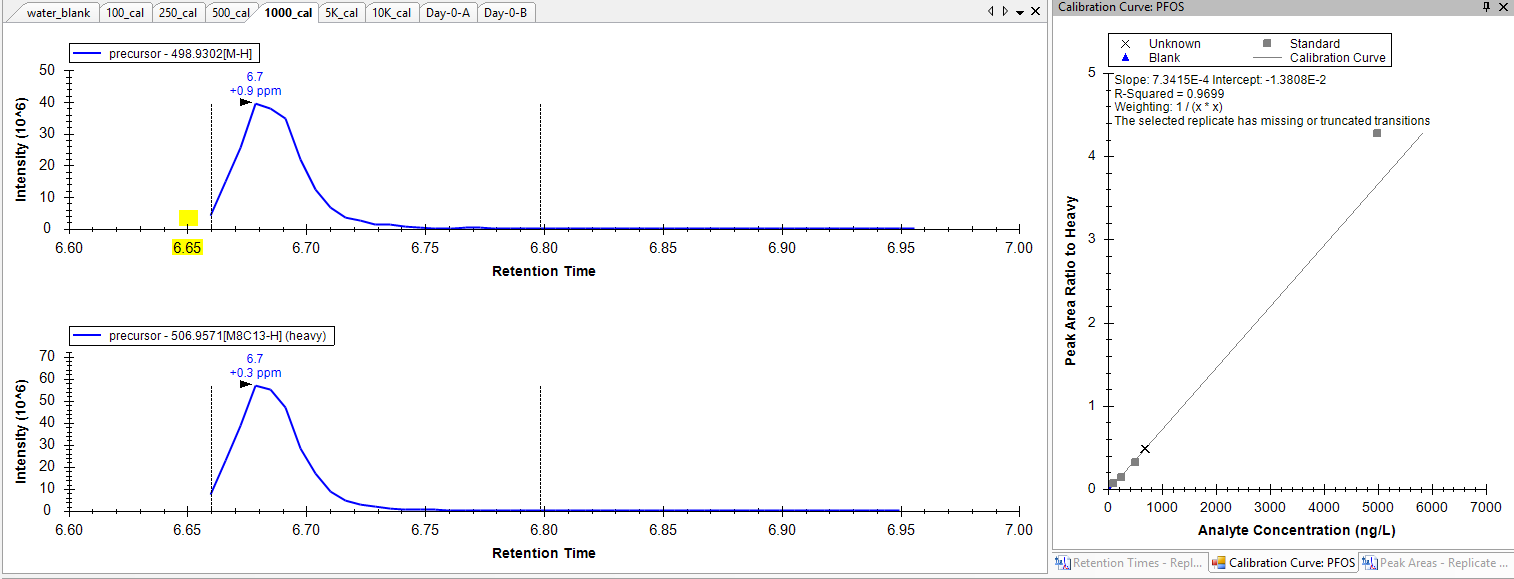
When I import the data into skyline I see all expected peaks but often times the leading edge of the peak is highly truncated. This truncation often throws an error which can prevent proper integration and calibration ("MS1.PNG") - "The selected replicate has missing or truncated transitions". I am able to "rescue" these peaks using a trick Brendan mentioned in a similar thread (by bumping the integration boundary inward), however, this truncation is an error as far as I can tell because when I interrogate the data in a vendor spectral viewer I can see that there is data that doesn't seem to make it into skyline. I have attached an image from the vendor software ("Truncated_Peak.PNG") which shows that this mass appears (within the mass tolerance) in the MS1 dimension at a RT that is not properly tracked by skyline in the MS1 dimension. I am thinking I may have some setting set too strictly which is causing the peak to not be picked up until part way through it's elution?
I have attached the skyline document .zip here. In this document I am quanting using both MS1 and MS2 data. Most of the truncation occurs in the MS1 dimension (see for example the 1000 cal point which throws an error). I also see some strange integration in the MS2 dimension ("MS2.PNG") where it appears skyline is trying to "connect" data points that are separated by a long stretch of time (see for example the 500 cal point) and therefore not properly allowing the peak to discontinue or return to baseline. This also appears to me to be related to peak truncation and an improper parsing of data that I believe is actually present in the raw file and which would make these peaks look more normal...
Thanks for any help you can provide!
|
| |
| Brian Pratt responded: |
2020-07-17 12:15 |
Can you provide one or two of the data files involved, so we can better understand the whole picture? You can upload them to http://skyline.ms/files.url or we can arrange something else if you prefer.
Thanks for using the Skyline support board,
Brian Pratt
|
| |
| Nick Shulman responded: |
2020-07-17 12:15 |
I think the bug here might be that the calibration curve is not supposed to care about truncated peaks if your normalization method is "ratio to heavy". Let me get back to you on this.
By the way, we have added a new feature in Skyline-Daily which might help with this scenario. On:
Settings > Transition Settings > Instrument
there is a new checkbox "triggered acquisition".
When Triggered Acquisition is selected, Skyline will notice when there are gaps in the MS2 scans for a transition, and it will excise those gaps from the MS1 chromatograms for that molecule.
In your Skyline document, I noticed that your MS1 transitions are under a different molecule than your MS2 transitions. In order for the Triggered Acquisition feature to work correctly, I think you would need to combine them under the same molecule.
Here is a page that talks about how this new feature works:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=TriggeredAcquisition
-- Nick |
| |
| jrenders responded: |
2020-07-17 12:51 |
Hi Brian,
I uploaded the data where you indicated.
Hi Nick,
I saw the "Triggered Acquisition" feature and thought for sure that would help me out, but I got an error every time I tried to use it. I've now tried as you indicated - added all transitions to the same molecule (see attached image), however, I get the following error upon attempted data import:
At 3:47 PM:
Failed importing results file 'D:\METRIC\JRE\Data\20200717_Buchwalter_Water_Sample_Redos_PFOS\20200717_Buchwalter_PFOS_2500_cal.raw'.
Object reference not set to an instance of an object.
pwiz.Skyline.Model.Results.ChromCacheBuildException: Failed importing results file 'D:\METRIC\JRE\Data\20200717_Buchwalter_Water_Sample_Redos_PFOS\20200717_Buchwalter_PFOS_2500_cal.raw'.
Object reference not set to an instance of an object. ---> System.NullReferenceException: Object reference not set to an instance of an object.
at pwiz.Skyline.Model.Results.SpectrumFilter.GetSureQuantScanDescription(SrmSettings settings, PeptideDocNode peptideDocNode, TransitionGroupDocNode transitionGroupDocNode) in C:\proj\pwiz_x64\pwiz_tools\Skyline\Model\Results\SpectrumFilter.cs:line 1319
at pwiz.Skyline.Model.Results.SpectrumFilter..ctor(SrmDocument document, MsDataFileUri msDataFileUri, IFilterInstrumentInfo instrumentInfo, Nullable`1 maxObservedIonMobilityValue, IRetentionTimePredictor retentionTimePredictor, Boolean firstPass, GlobalChromatogramExtractor gce) in C:\proj\pwiz_x64\pwiz_tools\Skyline\Model\Results\SpectrumFilter.cs:line 285
at pwiz.Skyline.Model.Results.SpectraChromDataProvider..ctor(MsDataFileImpl dataFile, ChromFileInfo fileInfo, SrmDocument document, IRetentionTimePredictor retentionTimePredictor, String cachePath, IProgressStatus status, Int32 startPercent, Int32 endPercent, IProgressMonitor loader) in C:\proj\pwiz_x64\pwiz_tools\Skyline\Model\Results\SpectraChromDataProvider.cs:line 121
at pwiz.Skyline.Model.Results.ChromCacheBuilder.CreateSpectraChromProvider(MsDataFileImpl dataFile, ChromFileInfo fileInfo) in C:\proj\pwiz_x64\pwiz_tools\Skyline\Model\Results\ChromCacheBuilder.cs:line 1264
at pwiz.Skyline.Model.Results.ChromCacheBuilder.BuildCache() in C:\proj\pwiz_x64\pwiz_tools\Skyline\Model\Results\ChromCacheBuilder.cs:line 233
--- End of inner exception stack trace ---
Open to any suggestions, thanks! |
|
| |
| jrenders responded: |
2020-07-31 09:20 |
Hi all,
Do you have any suggestions for getting skyline to recognize leading edges of MS1 peaks? I still seem to be getting some pretty significant truncation of my peaks despite there seemingly being good data there. I've attached another recent peak (viewed in skyline and Freestyle) showing the parts that seemed to be getting missed... |
|
| |
| Nick Shulman responded: |
2020-07-31 11:07 |
There is a bug Skyline-Daily which prevents "Triggered Acquisition" from working in small molecule documents. This will be fixed in the next update of Skyline-Daily.
Unfortunately, this does not fix your problem, since even with "Triggered Acquisition" enabled, the peaks still get marked as "truncated", and truncated peaks do not get used when calculating calibration curves.
I was surprised that truncated peaks do not get included in quantification (for calibration curves or for group comparisons). I had thought that truncated peaks would be included if you were doing ratio to heavy normalization, but that is not the case. If you are doing ratio to heavy the Skyline allows some transitions to have missing values or be truncated, and Skyline uses the rest of the non-truncated values for the calculation. If all of the transition peaks are truncated, then the calculated value equals null.
I am not sure what the behavior should be:
1. Always use truncated peaks if the normalization method is ratio to heavy.
2. Add an option somewhere to decide whether truncated peaks should be included.
3. Allow truncated peaks if "triggered acquisition" is selected.
I will start an email thread where we can discuss this.
-- Nick |
| |
| jrenders responded: |
2020-07-31 11:54 |
Thanks for the reply, Nick. Though I am concerned about truncated peaks getting used for calibration, I am also concerned about why the peak is truncated in the first place when it appears that this MS1 mass should produce a nice (fairly) Gaussian peak in MS1 space. This is what I was trying to confirm via Freestyle (Thermo's data browser) and what I showed in the attached image. How does skyline decide which data contribute to this MS1 trace? There is something that is causing it to miss the first 2-3 MS1 data points resulting in a truncated peak in a lot of my analytes. This causes the peak to drop from calibration, but even if it were included the peak area itself would be wrong because the peak is not complete. Is there a way for me to increase some tolerance to try to catch those leading peak edges? Thanks. |
| |
| Nick Shulman responded: |
2020-07-31 12:55 |
The reason that your MS1 chromatograms are getting truncated is that Skyline sees that you have MS1 and MS2 chromatograms for the same molecule, and wants to make them the same length.
Skyline does this because it is the right thing to do in the case of scheduled PRM methods.
In your case, you have put your precursor and product transitions under two different molecules in two different molecule list. However, the chromatogram code in Skyline does not know about molecule lists. That code believes that chromatograms come from the same precursor if their molecule name and precursor m/z match.
The easiest way to work around this behavior would be to give your molecules two different names.
Here's a document that I made where I renamed one of your molecules "PFOS_precursor" and then you get full length MS1 chromatograms.
-- Nick |
|
| |
| jrenders responded: |
2020-08-01 08:23 |
Nick,
!Brilliant! That's the bit I was missing! I'll play around more with how I group my precursors and transitions. I just saw that the triggered acquisition was fixed as well - I can confirm this has has successfully truncated the MS2 peaks appropriately. Thanks for your help with both of these issues! |
| |
|
|
 MS1.PNG
MS1.PNG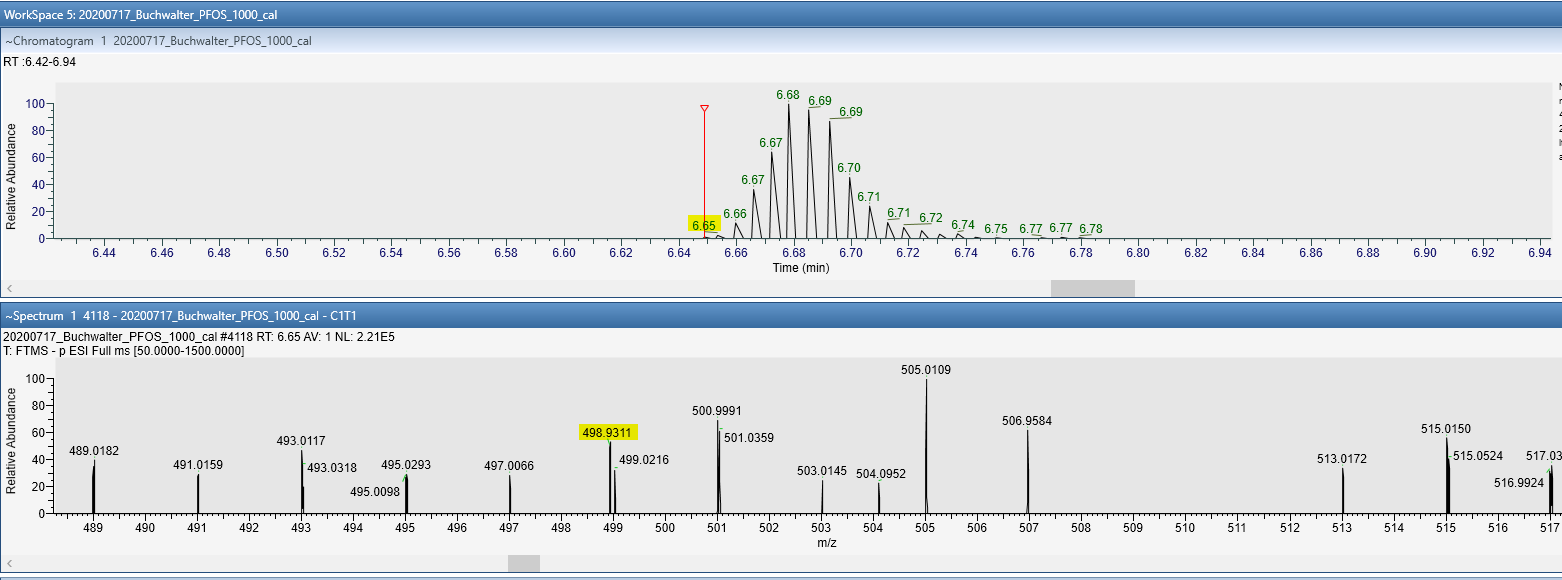 Truncated_Peak.PNG
Truncated_Peak.PNG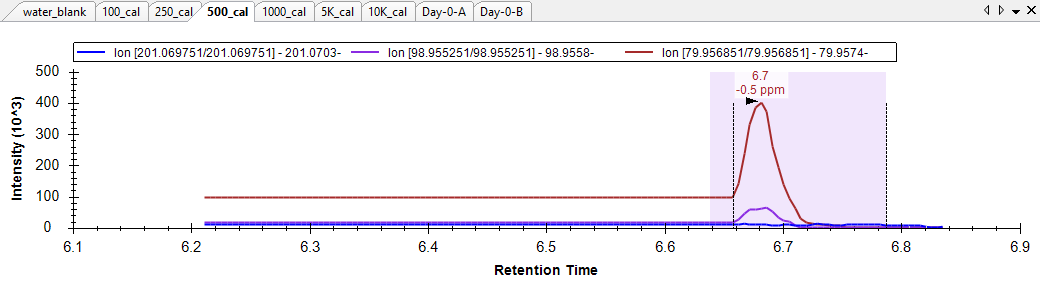 MS2.PNG
MS2.PNG Molecules.PNG
Molecules.PNG Picture2.png
Picture2.png