| Brian Pratt responded: |
2020-02-26 09:44 |
Hi Jonas,
You'll probably have to work out a way to use msconvert to create mzML equivalent files with one or the other spectra type filtered out.
Best,
Brian Pratt
|
| |
| Brendan MacLean responded: |
2020-02-26 10:13 |
Brian is right. You should use msconvert to filter your files into two different mzML files, one with HCD only and one with EThcD only.
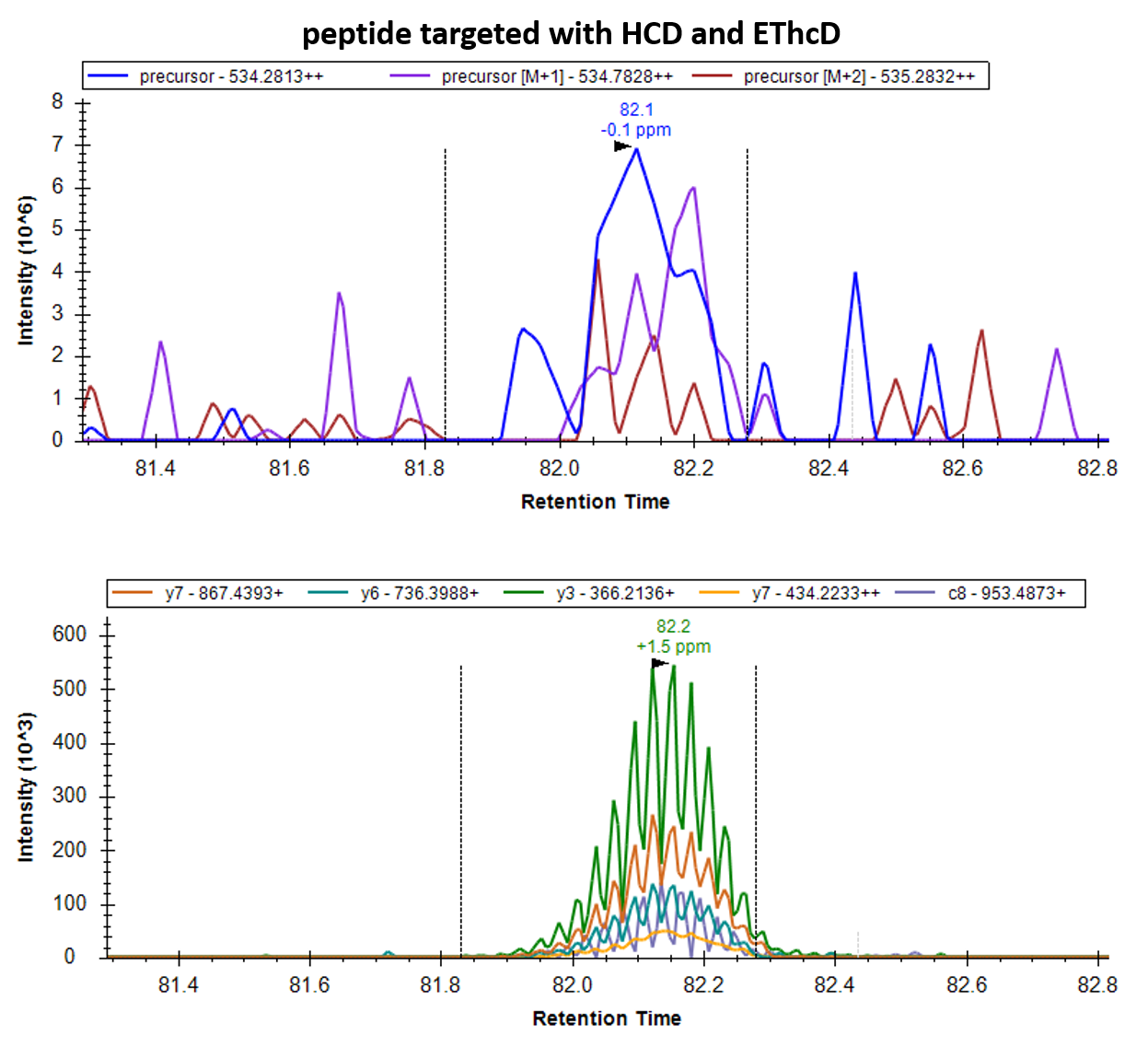
I like picture 1. You can even see which points come from HCD (higher intensity on the y-ions) and which come from EThcD (higher intensity on the c-ion and lower on the y-ions).
Good luck. Interesting idea.
|
| |
| jonasbecker responded: |
2020-02-27 09:56 |
Hey Brian & Brendan,
that worked like a charm for the HCD data and partly for the EThcD data, thanks for the great support!
For extracting the EThcD spectra using msconvert, what would you suggest best to set as "activation type"?
Best regards and many thanks again,
Jonas
|
| |
| Matt Chambers responded: |
2020-02-27 10:20 |
Hi Jonas,
Filter on ETD. ETD is the primary mode for any ETD with supplemental activation; the filter is based on the primary mode.
|
| |
| jonasbecker responded: |
2020-02-27 11:39 |
Perfect! Thanks again for the quick response!
|
| |
| jonasbecker responded: |
2020-03-03 09:57 |
Dear all,
I used Skyline now to combine the two files again since my standard peptides were measured only with HCD while the peptides of interest were measured using EThcD.
Now I would like to use the standards peptides as "global standard" however, Skyline can't perform this action since the data is coming from different mzml files.
Is there any chance to circumvent this issue?
Best,
Jonas
|
| |
| Brendan MacLean responded: |
2020-03-03 11:24 |
Hi Jonas,
So is this PRM? If so, you can also use MSConvert to filter by precursor m/z. I have used this to filter out DIA/SWATH isolation windows which were incorrectly placed. It might be a bit cumbersome, but it is about all I can think of for what you are describing.
--Brendan
|
| |
| jonasbecker responded: |
2020-03-04 04:18 |
Hi Brendan,
yes, it is PRM data. I think the problem would still be that I have a list of HCD-fragmented standard peptides plus a list of EThcD-fragmented peptides of interest which should be in the same mzML file so I can use the standard peptides as "global" to normalize. Unfortunately, I'm not sure on how to combine the activation type & precourser m/z filter to get all spectra in one file. Is it even possible?
Thanks for your help,
Jonas
|
| |
| Matt Chambers responded: |
2020-03-04 07:34 |
Hi Jonas,
One possibility would be to manually search & replace the activation type in the converted file so they are all the same. That would be pretty hacky, and it would be easier in mzXML than mzML.
It's also plausible to add this capability to msconvert directly (it not currently possible), but I hesitate to enable such a hacky workaround. What do you think Brendan?
|
| |
 picture1.png
picture1.png picture2.png
picture2.png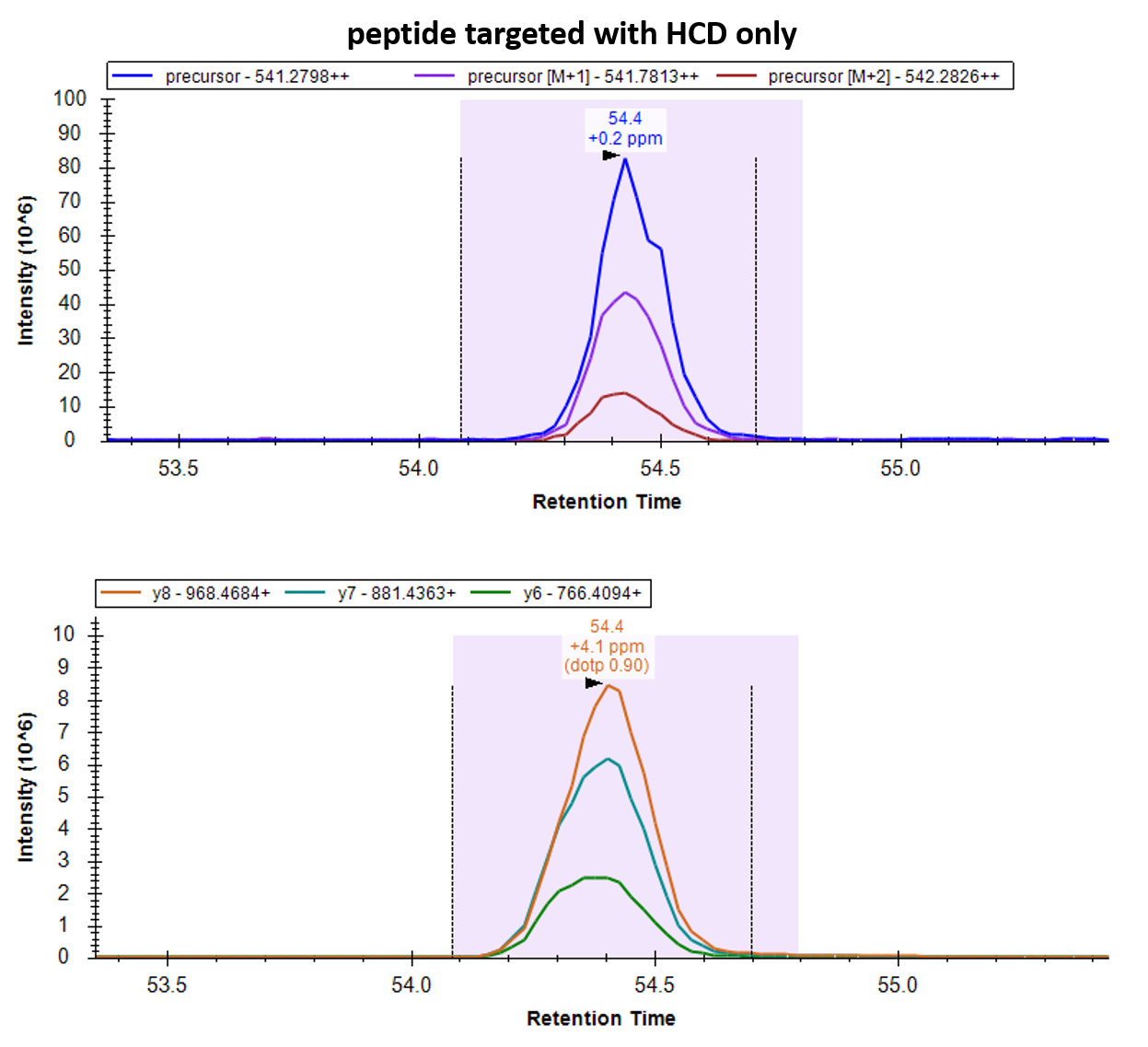 picture3.png
picture3.png