| Nick Shulman responded: |
2020-01-16 10:54 |
Can you send us your Skyline document?
In Skyline you can use the menu item:
File > Share
to create a zip file containing your Skyline document and supporting files including extracted chromatograms, spectral libraries and iRT database.
If that .zip file is less than 50MB then you can attach it to this support request.
Otherwise, you can upload it here:
https://skyline.ms/files.url
-- Nick |
| |
| romeally responded: |
2020-01-16 11:33 |
Thanks...here is the zipped share. I had run the iRT standards alone and then spiked in. They seem reproducible however the actual peptides have some that elute earlier and later than the first and last iRT standards. I have been using skyline for some time but I am just starting to add iRTs to my larger assays. Appreciated! Bob |
|
| |
| romeally responded: |
2020-01-16 12:47 |
I think I figured it out. I had made 3 methods but acquired with the same method 3 times in my schedule. I am missing values for the second 2 calibration runs. They all contain precursor data, but the second 2 have no fragment ion data for their corresponding precursors. I am rerunning...hopefully this will work better! |
| |
| Nick Shulman responded: |
2020-01-16 13:11 |
Bob,
Skyline has a window which is very useful for figuring out what is going on with these sorts of "Regression Failed" problems.
You can bring up the Retention Times Linear Regression window by using the menu item:
View > Retention Times > Regression > Score To Run
Then, on the linear regression graph there are a bunch of things you need to choose for it to be useful:
Calculator > iRT_Pierce_C18Heavy
Points > Standards
If you do that, then you see a graph that looks like picture that I have attached. You can see that the times of most of your standards are ending up very far from the straight line. This is happening because Skyline is choosing the wrong peak for most of these standards in your document.
I believe the reason that Skyline is choosing the wrong peaks is that you have told Skyline to look for all of the B and Y ions. Skyline does not know which of those transitions are likely to be detectable, and so Skyline is choosing peaks that probably came from other peptides.
If you delete your iRT standards from the Targets tree, you can get Skyline to add them back with our recommended set of transitions.
That is, do the following:
1. Delete the Peptide List "Pierce Peptides" from the Targets tree.
2. Settings > Peptide Settings > Prediction and choose "None" in the Retention Time Predictor dropdown.
3. Press OK on the Peptide Settings dialog
4. Settings > Peptide Settings > Prediction and choose "iRT_Pierce_C18Heavy" in the Retention Time Predictor
5. Press OK on the Peptide Settings dialog.
Skyline will then offer to add the iRT standards to your document. Skyline will ask you how many transitions you want per peptide. I like to choose 6.
After this, you need to tell Skyline to do the peak finding again using this new set of transitions.
To do that, you go to:
Edit > Manage Results > Rescore
After you do all that, you will end up with a much better looking linear regression window.
After I did all that, I was still not able to use the "Use Results" button in the iRT calculator dialog.
I think I will need to see your .raw files in order to figure out what the final problem is preventing the linear regression from working. The Linear Regression window in Skyline does not do a good job when you have multiple files per replicate. The graph averages numbers together and so it's really hard to see whether individual files within the replicate are having problems.
I hope this helps.
-- Nick |
|
| |
| romeally responded: |
2020-01-17 10:11 |
So I did what you said and Skyline never asked to add the iRT standards into my document. I tried reimporting the data and now I see no chromatograms at all. So now I have a document with no iRT standards and although it says it imported my data I see no chromatograms. So this is PRM data and I added a library. I don't think Skyline is looking for B ions but maybe I am wrong. The chromatograms from the data looked pretty good before I deleted the iRT peptides from the table. If I run the iRT standards alone they look very good. Perhaps I am not spiking enough in? I will share the current document...I don't know where all my chromatograms went and I don't see the individual runs I selected to import. This should be 3 injections of the newly acquired data. |
| |
| romeally responded: |
2020-01-17 10:13 |
Ok I can't share the document. It says it has to be fully loaded to share, however I imported 10 minutes ago and I see nothing coming in. It thinks it is still loading but it is just stuck or something. I am going to try rerunning the samples with a larger spike. I will share the standards alone skyline document. |
| |
| romeally responded: |
2020-01-17 10:15 |
Here are the iRT standards alone...no issues with this only when spiked in. Not sure what the problem is... |
|
| |
| Nick Shulman responded: |
2020-01-17 14:05 |
Bob,
The instructions I gave you about deleting your iRT peptides and then getting Skyline to add them again by going into Peptide Settings only actually works in Skyline-Daily. In Skyline 19.1 I could not figure out how to get Skyline to add the peptides to your document.
Skyline-Daily seems to have a different bug which completely prevents the "Use Results" button from working. We are going to try to fix that.
I have attached a copy of your Skyline document where I have changed the set of transitions on your iRT standards.
Skyline is able to correctly identify the peaks and so when you go to:
Settings > Peptide Settings > Prediction > Calculator Button > Edit Current > Add > Add Results
everything works great (using Skyline 19.1).
Let me know if you have any new questions.
-- Nick |
|
| |
| romeally responded: |
2020-01-18 06:21 |
Thanks Nick! So I reran everything and it appears to be working. I am still finding some things that don't completely make sense. So I told the document to only take y ions but when I look at my peptides I still see b ions in the sample peptides but not the iRT. I told it to pick 6 product ions from the filter but I still see more than 6. This probably hinders my scoring which leaves me with few green hits. It always looks so nice with MRM data since you pick the transitions and they are the only ones found or possible, but with PRM data there is a long list of possible product ions. So now I am ready to export this as a scheduled single run. If I wish to change my gradient how will this affect the retention times? Will the method use the unitless iRT values or will it recalculate the retention times? The .csv will have start and stop times adjusted? I was under the assumption that once I got this right I can change columns or gradients and should not have to perform these steps again. I now have it accepting the entire list of peptides into the iRT. I assume I should save this as a separate iRT and keep the initial Pierce iRT alone as a reference cal? So I should be able to use the initial iRT cal in other methods with different peptides so long as I redo the Skyline precursor list and perform these steps again? I think I got it I just need to crystallize the entire concept in my brain and explain the things that are different than I expected like the b ions presence and the more than 6 product ions when I told it only y and 6. I attached my latest Skyline document. Thanks again! Bob |
|
| |
| romeally responded: |
2020-01-19 05:04 |
So I ran a scheduled run with the iRT calibration and gave the peptides a 5 minute window but I have some that are withing one minute of the predicted retention time but still got cut off. I am confused...shouldn't I have gotten everything within 5 minutes of the predicted retention time? I can see a tick that says predicted one or two minutes away and yet the peak is still cut off. attaching latest skyline documents of scheduled run post iRT calibration. |
|
| |
| Nick Shulman responded: |
2020-01-19 16:31 |
Bob,
Your iRT standard peptide GISNEGQNASIK was found at different times in your different injections in 224_Peptides_+HeavyPierceC18iRT_Edit200118.sky.zip
In two of the injections, that peptide was found where it was supposed to be at it's predicted time of 11.9 minutes. However, in your injection "...CalibrationSet3_C.raw", it was found at 37.0 minutes.
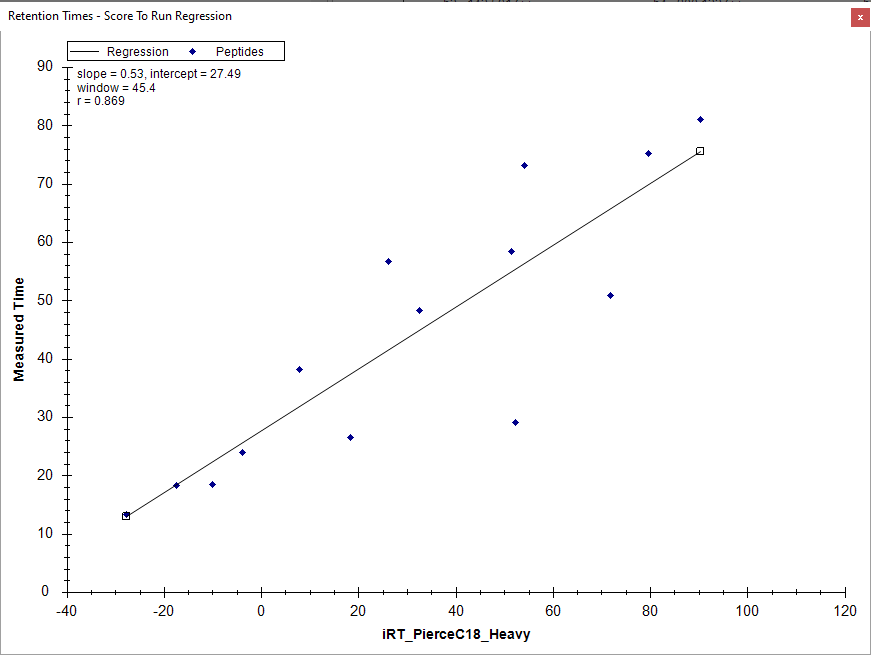
When you look at the Retention Time Linear Regression graph pane, Skyline plots that peptide at the average of those three numbers (20.3 minutes), and in the attached picture you can see that it is an outlier.
Because that iRT standard peptide was not found at the correct time in that particular injection, it means that the predicted retention times are going to be wrong for all of the peptides that you measured in that particular injection.
In your file "224_Peptides_+HeavyPierceC18iRT_ScheduledRun_5minutes.sky.zip" I think you will find that the peptides with truncated chromatograms (such as "SQYLQLGPSR") are all peptides that were measured in "...CalibrationSet3_C.raw"
I think that if you manually choose the correct peak for GISNEGQNASIK in "...CalibrationSet3_C.raw" and then export a scheduled method it will have the correct times in it.
Hope this helps,
-- Nick |
|
| |
| romeally responded: |
2020-01-21 09:20 |
Ok now I see it. So I suppose if I can't determine any one peptide in the iRT it is best to remove outliers prior to setting the calibration. So when I add the peptides I wish to measure to the iRT calibration I noticed that the it did not ask me to save the calibration with the iRT plus peptides. Is the iRT calibration changed when I add the new peptides? Is this only changed in the current open document? If I wish to use the iRT calibration with different sets of peptides or with a different gradient do I need to recalibrate the iRT mix? I am also still confused when I told my document to only look for 6 ions from the filter and only y-ions why I get more than 6 and b-ions are included. With MRM data this is clean since you are only picking certain transistions, but with PRM data there are many more possibilities and some are cleaner ions than others. Thanks for your help...I think this is working within its own framework but I want to be able to transfer this calibration to different columns and gradients. What do I need to do to transfer these known iRT values into different conditions? |
| |
 LinearRegression.png
LinearRegression.png GISNEGQNASIK.png
GISNEGQNASIK.png