| Nick Shulman responded: |
2020-01-08 11:06 |
Fynn,
Can you send us one of your .raw files (e.g. "20200106_EXPL2_FyHa_SA_PRM_BSA_01.raw")?
You can upload that file here:
https://skyline.ms/files.url
Usually bugs like this have been caused by Skyline being confused about what m/z was isolated in each MS2 scan and so the scans never get properly associated with the peptides in your document.
-- Nick |
| |
| Matt Chambers responded: |
2020-01-08 11:52 |
Even better would be one RAW file from each instrument so we can see the difference. |
| |
| Fynn responded: |
2020-01-09 00:48 |
Hi all,
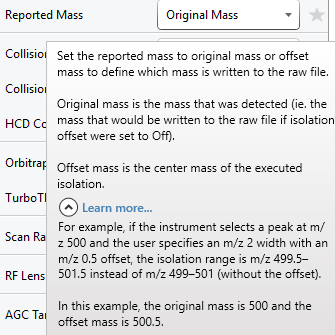
I figured out why the import was not working. It was a problem with the Exploris settings after all. The new Xcalibur version has an option called "Reported mass" when you define an isolation offset. This option was set by default to "offset mass", which leads to the recording of an offset mass and not the original mass (see picture). After changing this point to "original mass" the import of the file into skyline works. I am very sorry for the confusion.
Best,
Fynn |
|
| |
| Brendan MacLean responded: |
2020-01-09 07:47 |
We should still get your data - both types. (please) From my read of the description you provided, I believe we would prefer the Offset mass, i.e. the central mass at which the quadrupole was set, and not the Original mass which I read as an instrument detected mass pulled from an MS1 spectrum that is likely the monoisotopic mass of the most abundant precursor in the isolated range. That is not really what we are expecting. So, it is a bit strange that this works better than the Offset mass.
We really need to take a closer look at how Thermo is representing these two masses. Thanks for bringing this to our attention.
--Brendan |
| |
| Matt Chambers responded: |
2020-01-13 10:25 |
The offset mass may be the central mass at which the quadrupole was set, but the offset is a constant 0.4 m/z. So if using a narrow extraction range around each isotope then we really need the original mass because the m/z after offset won't actually correspond with a peak. Like this:

If using a wide window instead of multiple narrow windows, then the offset mass would be better. |
|
| |
 Screenshot (1).png
Screenshot (1).png