Dear Skyline team,
I am trying to extract MS1 quantification from timsDIA PASEF data. I have 4 raw files (.d folders) that were processed using FragPipe/MSFragger/Philosopher. Which, by the way, works great for these data.
I am not able to build a spectral library using the DDA-MS1 quant workflow, with Import - Peptide results etc for building the list form the search results. Neither using MSFragger pepXML files nor using interact-.pep.xml files after PeptideProphet work. When using interact-.pep.xml files, I get an error that it cannot find spectral files. When using pepXML, it finds the files to get the spectra (from file_calibrated.mgf files produced by MSFragger) but gives an error at a later stage.
However, I am able to trick Skyline into extracting MS1 quant from DDA files by going through the DIA workflow. When going that route, I am able to select pepXML files, Skyline finds the corresponding spectral files, and it can build the library. I can also select .d DDA files and perform quantification. However, with this trick, Skyline tries to quantify both precursor and y/b ions (because it is a DIA workflow), which takes a lot of time. I just need precursor quantification from MS1, but removing b,y ions from the list of ions to quantify gives me an error.
Also not that using interact-*.pep.xml files (instead of pepXML from MSFragger) still does not work when using the DIA workflow.
So, ideally, I would like to being able to
- Load PeptideProphet pep.xml files, and not just the MSFragger pepXML files.
- Being able to use DDA-MS1 quant route rather than DIA route when working with DDA data.
Admittedly, I am an inexperienced Skyline user. So I may be doing something wrong. However, Brett Phinney seemed to confirm the errors I am getting with DDA files.
I could get someone in my team to work directly with the Skyline team to resolve these issues, or to learn how to do this analysis properly.
Thank you,
Alexey Nesvizhskii
University of Michigan
|
| |
| Brendan MacLean responded: |
2019-12-13 08:24 |
Hi Alexey,
Really appreciate your pushing this forward with your feedback. It sounds like maybe the problem is just in the naming?
<file basename>_calibrated.mgf
Is that right? I think you could probably get away with <file basename>.calibrated.mgf, because we already built support for the Aebersold lab extracting search spectra to <file basename>.c.mzXML (the .c for centroided). It sounds to me like the problem is in Skyline trying to match the spectrum source file naming to the chromatogram extraction file names, which will be critical to having match ID times and run-to-run alignment, clickable ID anotations, etc.
So, I really think we want this DDA search spectrum source file to chromatogram extraction file connection, rather than using the DIA workflow, which explicitly expects no connection between the searched DDA (library building) files and the extraction DIA files.
Can you post screenshots of what you are seeing for "errors"? Do I have this right? I am happy to jump on a screen sharing meeting with you and go over this today.
--Brendan
|
| |
| nesvi responded: |
2019-12-13 08:37 |
Hi Brendan,
I think if I can show you on my screen it would be the easiest. I will check if renaming to <file basename>.calibrated.mgf makes a difference.
What do you prefer for screen sharing? If you have it set up, maybe you can send me an invitation directly. I can show you how the folders are structured and what I am doing to load the data.
Best,
Alexey
|
| |
| nesvi responded: |
2019-12-13 08:55 |
To prepare for the call, here is what happens when I use DDA workflow, Import Peptide search results - select <basename>.pepXML file (Screenshot 1). I am not able to use Find and select .d folder
If I select interact-<basename>.pep.xml I get an error at the first step (Screenshot 2). It looks like it is not even looking for _calibrated.mgf in this case. Only mzML, mzXML etc, but not mgf
Changing to from _calibrated to .calibrate did not help (when using pepXML, it was still searching for _calibrated.mgf and cound not find it).
Thanks
Alexey
|
|
| |
| nesvi responded: |
2019-12-13 09:36 |
I uploaded a zipped file containing MSFragger pepXML file, the corresponding _calibrated.mgf file, and interact-*.pep.xml file after PeptideProphet
The file name is MSFragger_pepxml.zip
|
| |
| Brendan MacLean responded: |
2019-12-13 09:45 |
Thanks, Alexey. We will have a look, and I am sure we can have this working for the next Skyline-daily release.
|
| |
| nesvi responded: |
2019-12-16 09:29 |
Dear Skyline team,
Thank you so much for making the changes. The new Skyline-daily fixed the issues above.
I have a few additional questions.
- I want to confirm that you are parsing the ion mobility information from interact.pep.xml file.
The tag is "ion_mobility"
e.g.
<spectrum_query start_scan="99780" ion_mobility="1.3496233" uncalibrated_precursor_neutral_mass="822.41187" assumed_charge="1" spectrum="20180819_TIMS2_12-2_AnBr_SA_200ng_HeLa_50cm_120min_100ms_11CT_1_A1_01_2767.99780.99780.1" end_scan="99780" index="31820" precursor_neutral_mass="822.4133" retention_time_sec="2065.148">
I do not see the ion mobility value shown anywhere in the library.
When tracing MS1 to get XIC, are you restricting the data in the ion mobility dimension?
Ideally, you would want to trace only MS1 signals in the correspond IM window, MS2 scan ion mobility +- some Delta.
In the example above, 1.3496233 +- e.g. 0.05
- I see a parameter in the XIC tracing window settings (see attached screenshot)
"Use spectral library ion mobility values when present"
Resolving power: 30
So, if you do read the ion mobility value, and if this is the parameter that tells Skyline to use it during the MS1 XIC tracing step, how do you define the resolving power here?
The IM values we work with are on the Bruker's 1/k0 scale, with values ~ 1 (as in the example above). When we trace peaks in our IMQuant tool, we use +-0.05. So I am trying to understand how to convert that 'resolving power:30' to the window +- 0.05 around the ion mobility value we write in the MS2 scan line in pep.xml.
Thanks!
Alexey
|
|
| |
| Brendan MacLean responded: |
2019-12-16 10:17 |
Hi Alexey,
Thanks for confirming the fix and pushing forward with your testing.
- I checked the code in PepXmlReader before releasing and found: ionMobility = getDoubleAttrValueOr("ion_mobility", attr, 0.0); The fact that you see the Ion mobility filtering group box implies that your .blib file contains ion mobiltiy information. I see the RefSpectra.ionMobility column populated in the .blib I built from your pepXML. When you use View > Spectral Libraries to look at the library, you should see IM values with RT values just above the Close button e.g. "RT: 50.15 IM: 1.2100Vs/cm^2" (from a PASEF library on my computer)
- Yes, that is what is meant by use IM values from the library when present. The extraction width (+/-) is determined by the resolving power you specify (in this case 30). For diaPASEF, I have found 50 resolving power to work well, but anywhere between 20-50 is probably fine. You can check that this is happening for sure, but clicking on a chromatogram in a chromatogram graph and checking the "Full-Scan" spectrum view (as described in the Ion Mobility Filtering tutorial - https://skyline.ms/_webdav/home/software/Skyline/@files/tutorials/IMSFiltering-3_7.pdf#page=12 - worth a read, even though it uses Agilent IMS data). If IMS filtering is being applied, you will see a horizontal magenta filtering band in the plot. You can also access the IMS filtering values in Skyline reports as: Ion Mobility MS1, Ion Mobility Fragment, Ion Mobility Window.
- I think resolving power is a general unit independent concept which normally means FWHM (a.k.a resolution) = (value / rp). So, a value of 1.2 / 30 = 0.04, which means 25 rp would be closer to your goal of 0.05, because we are using 2 x resolution as the extraction window. Checking this in diaPASEF data I have processed with 20 rp, I see IM value = 0.92, IM window = 0.09 - a very simple calculation to check, since 20 rp should simply shift the value one decimal place right as it does above) This is how Erin Baker wanted us to do things when we started working with IMS, and since it fit well with what we do in the m/z dimension, we were happy to oblige.
Hope this helps to clarify. Let us know what you see, if you are not getting IMS filtering or IMS values in View > Spectral Libraries, we will want to look at your files again.
Thanks again for the testing and feedback.
--Brendan
|
| |
| Brendan MacLean responded: |
2019-12-16 10:23 |
P.S. - I also see the IM value type in the .blib file I built from your pepXML correctly assigned. You can open .blib files yourself with SQLite Expert (or similar) and see:
- IonMobilityTypes:
id ionMobilityType
0 none
1 driftTime(msec)
2 inverseK0(Vsec/cm^2)
3 compensation(V)
- RefSpectra:
id peptideSeq precursorMZ precursorCharge peptideModSeq ionMobility collisionalCrossSectionSqA ionMobilityHighEnergyOffset ionMobilityType ...
1 EMLQQSK 879.42561499433 1 EM[+15.9949]LQQSK 1.3543512 0 0 2
2 KPAAAAAPGTAEK 591.826930001708 2 KPAAAAAPGTAEK 0.93656766 0 0 2
But, it is much simpler to check in the View > Spectral Libraries viewer.
|
| |
| nesvi responded: |
2019-12-16 14:22 |
Dear Brendan,
I checked and yes IM info is used, and the setting of 30 seem reasonable for the IM resolution, and everything looks fine so far.
Thanks,
Alexey
|
| |
|
|
 Screenshot2.png
Screenshot2.png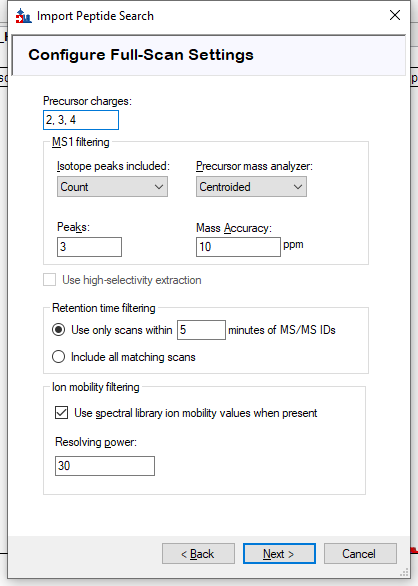 Screenshot 2019-12-16 12.05.10.png
Screenshot 2019-12-16 12.05.10.png