| |
| Brendan MacLean responded: |
2019-11-29 10:55 |
Skyline should be making pretty good use of parallelism at this point. We have spent a lot of time on it. But the parallel functions of Skyline are mostly limited multi-file parallelism, which is controlled by the choice you make a file import time of "Files to import simultaneously" in the "Import Results" form, which has defaulted to "One at a time", but which I just changed to default to "Many", the setting I would definitely recommend on a Dell PowerEdge (at least with the current Skyline-daily, but also with just 2 files). Also, --import-threads=# at the command-line should be even better, or --import-process-count=# for Skyline 19.1 (which has better multi-process performance than multi-threaded).
That said, I am not a huge fan of just building bigger and bigger lists of targets. If the library contains 581,197 precursors, then likely you have used the default of 581,197 precursors for decoys. Though, even with the much smaller Pan Human spectral library from ETH, I felt using 1/4 as many decoys was sufficient and had a performance benefit.
Assuming you have doubled your numbers with decoys, then you would have 1,122,934 precursors, which even with just 6 fragment ions each would mean 6.1 million transitions. If you included precursor transitions extracted from MS1 spectra, then you likely doubled the amount of chromatogram extraction that Skyline does.
For just 2 DIA files on the system you have, this should still work out okay, but the current Skyline implementation would limit you to 2 threads for spectrum retrieval (which would be relatively inactive), 2 threads for chromatogram extractions, and 8 threads for peak scoring. So, it is unlikely to make full use of a Dell Xeon PowerEdge with... how many cores? 24, i.e. 48 logical processors after hyperthreading?
For this, you really need to increase the file count to at least 6. Though, I hope you don't want to increase this many targets to large numbers of biologically important smaples. I would definitely recommend starting with some replicate (more than 2, though) refinement around what you can hope to detect reliably and what you can hope to measure with low enough technical variance to hope to see important biological variance.
Anyway, it should be possible to at least import your two files in parallel, and you should get better performance out of Skyline-daily right now than out of Skyline 19.1. We made some multithreading performance improvements in the past month or so to facilitate new diaPASEF support.
--Brendan
P.S. - Yes, probably your exact scenario would benefit from our allowing higher single-file parallelism at the chromatogram extraction level, given the small number of files and the massive number of transitions and the number of cores on your computer, but we have not yet built that flexibility.
|
| |
| Chinmaya k responded: |
2019-12-09 10:53 |
Hi Brendan,
Thank you for the response.
Finally, I was able to finish the DIA data search for 2 .raw files against the spectral library as mentioned earlier. However, the system took more than 4 days to extract and import both MS1 and MS/MS information and match it with the respective spectral library. As you have mentioned, I have set the import options to several files at a time for maximum system utilization and it did work out by occupying 100% of both 128 GB RAM and >1.5 TB disk space (There were some Proteome Discoverer searches were also performed in between).
The library which I have used consists of 290,000 precursors corresponding to 239,154 peptides, which in turn with an equal number of decoys got increased to 581,197 precursors corresponding to 478,304 respectively as mentioned above. How does the usage of 1/4th of decoys help in calculating FDR ? can you please elaborate?
I had 2,55,61,964 transitions with decoys.
The system has 22 cores with 44 logical processors and yes, the slowness was observed in spectrum retrieval when compared to other steps.
However, a new Skyline-daily gets hang when I tried to reopen transition setting once spectral library match and decoy generation was performed with the same set of spectral library and could not continue thereafter.
Thank you,
Chinmaya
|
| |
| Brendan MacLean responded: |
2019-12-09 11:24 |
I can't quite read that number "2,55,61,964". The thousands separators seem misplaced. The pan-human assay which I have definitely processed a lot on a similar system without anything like a 4-day wait has 205,233 precursors and 163,053 peptides without decoys. When I was processing it a lot, I found that I got similar results using less than 1:1 targets:decoys. To test this I created a Skyline document with no decoys and used the command-line arguments (--decoys-add=shuffle --decoys-add-count=%DECOYS%) to vary the decoy count across a range of 2x values (or incrementing by log2) and I eventually settled on "40761" or 1/4 the number of peptides.
Skyline uses statistics (Storey-Tibshirani 2003) which do not require the targets:decoys ratio to be 1:1.
I guess there must just be an extra number in your transition total, and you are using close to 2.5 million transitions. That doesn't seem so bad to me, especially on the kind of system you are using, unless for some reason you are ending up doing full-gradient extraction, i.e. extracting all of your chromatograms across the entire gradient.
Thinking about this more, I think this must actually be what is happening to you. Can you please send a screenshot of your Transition Settings - Full-Scan tab and also explain how you are limiting chromatogram extraction to a subset of your gradient for each peptide? I really can't imagine anything like what you are reporting without somehow ending up extracting full-gradient chromatograms, which just isn't going to work for multiple reasons on this kind of analysis.
What you have described sounds only slightly larger than the Pan-Human assay, which I know works in Skyline on the type of system you have. So, once we get your settings worked out, this should be possible.
--Brendan
|
| |
| Chinmaya k responded: |
2019-12-10 02:07 |
Sorry for that, the comma separator above was applied as per Indian numbering system. However, there were approx. 25 million transitions in the skyline document (12,714,927/25,561,964).
For me, the use of 1/4 the number of decoy peptides is quite knew and thank you for letting us know. I will work on that aspect by varying the decoys as well using the same library and let you know.
As I have mentioned earlier, the transition settings consists of both MS1 and MS/MS filtering with 5 min predicted RT window under Retention time filtering section (Please find the attached ppt having screenshots of all the tabs under transition settings). I believe that I am trying to extract all chromatograms across the entire gradient (i.e. 140 min) and skyline does this sort of chromatogram extraction all the time I guess. Nevertheless, I have not seen any parameters to restrict chromatogram extraction across the entire gradient.
This is bit confusing about chromatogram extraction here for me. As you can see in the attachment, I have set the acquisition method to DIA with Isolation scheme set by importing the information's from .raw file ( It is 400-1000 m/z range with 10 Da isolation window with 0.5 Da margine) under MS/MS filtering with the resolution of 15,000 at 200 m/z. The search for scan around 5min of predicted RT window should help in minimizing random chromatogram extraction isn't it? This all I know about chomatogram extraction, please let me know if I am missing anything here.
Let me know if you need more info.
-- Chinmaya
|
|
| |
| Brendan MacLean responded: |
2019-12-10 08:36 |
So, 25,000,000 transitions for 500,000 precursors would imply that each precursor has on average 50 transitions, which is a very bad start.
I see the problem in your settings. You do not want to use "From filtered ion charges and types plus filtered product ions". This will give you very permissive transition selection and essentially select all possible charge 1 and 2, b- and y-ions for every precursor from y3/b3 to y[n-3]/b[n-3]. I think you want to change this to "From filtered product ions", and I also think you want to change your Transition Settings - Filter tab Product ion selection - To = "Last ion". It rarely makes any sense to use "Last ion - 1" or "Last ion - 2" and I have been considering removing these options. Perhaps you were desperate for a way to reduce the number of transitions.
Anyway, start with those 2 changes:
- Delete you decoys
- Transition Settings - Library - "From filtered product ions"
- Transition Settings - Filter - "Last ion"
- Regenerate your decoys
Then report back how many transitions this leaves you with. You should end up with fewer precursors because some will not support 6 transitions from their spectra any longer. And you should see all precursors with just 9 transitions. So, even if you have 500,000 precursors, you should only have 4.5 million transitions.
And this should import a lot quicker. Can you provide a screenshot of some of the chromatograms in your existing document to ensure they are showing only 10 minutes of chromatogram extracted, as you have indicated was your intention.
Progress...
|
| |
| Chinmaya k responded: |
2019-12-28 02:59 |
Hi Brendan,
Sorry for the late reply.
Thank you for all your previous suggestions. As you have mentioned it did work out. Now, the number of transitions, precursors, and peptides have reduced significantly to 46,21,343 trans/ 5,84,934 precursors/ 4,78,304 Peptides. The chromatogram extraction time from a single DIA raw file reduced from >24h to >10h.
Please find the attached screenshots of extracted chromatograms.
Recently, I have also faced issues to do reintegration by training mProphet model. Where it was showing a quite low number of target peaks (>20) available for a large number (>2,00,000) of decoy targets. What might be the reason for this error though there was a good number of target spectrum matches available?
Apart from this, I have observed Library intensity dot product was not being considered while mProphet model was trained since a few thousands of decoy peptides were not having any spectrum matches. Why mProphet or Skyline is not omitting such decoy peptides having zero matches?
As you have mentioned earlier in this thread, how about adding only 1/4th decoy peptides for the search? Does this help in reducing or completely removing decoy peptides with zero spectrum matches?
Thank you,
Chinmaya
|
|
| |
| Nick Shulman responded: |
2019-12-28 07:17 |
I am not sure that I understand your questions.
What do you mean by "quite a low number of target peaks available for a large number of decoy targets"? Can you post a screenshot of what you mean?
Is there something in particular that you want us to notice about your chromatogram screenshots?
When you train an mProphet model, Skyline can only use features that have scores available for all of the peptides in your document. If a particular peptide does not have a library dot product, then Skyline cannot use "library dot product" as a feature in the mProphet model. This is because Skyline is trying to generate a model which applies to all of the peptides in your document. If you would like Skyline to use the library dot product score then you need to delete all of the peptides from your document that are missing that score.
When you generate decoys in a Skyline document, those decoys remember which "real peptide" they were generated from. Whenever the decoy peptide needs a score associated with the spectral library, the decoy peptide uses the library spectrum belonging to the real peptide it was generated from. If you are seeing that decoy peptides are missing their spectral library information, then it should mean that the real peptide also is missing spectral library information.
If all of the real peptides in your document have spectral library information, but some decoy peptides are missing it, then it means that your decoys are out of sync with your targets. You should use the "Generate Decoys" menu item again to generate a new set of decoys where every decoy is associated with a peptide that is still in your Skyline document.
Another thing that can prevent a peptide from having a library dot product score is if the peptide has fewer than 3 MS2 transitions.
In the Edit Peak Scoring Model dialog you can see the distribution of feature scores on the "Feature Scores" tab. If some but not all of the peptides are missing a particular score then a binoculars icon will appear when you hover the mouse below the "Unknown" bar in the graph. Clicking that icon will cause Skyline to display the peptides that are missing that score in the "Find Results" window.
Hope this helps,
-- Nick |
| |
| Chinmaya k responded: |
2019-12-29 07:19 |
Hi Nick,
Please find the attached screenshot of the same error.
Where, both MS1 and MS2 chromatograms were extracted with 5 product ions or minimum 3. Only 1/2 decoy peptides were taken and removed peptides having 6 transitions (Refine > Advanced > Min transitions per precursor) post importing the results. I have also attached the Feature Scores window, where I do not see any "Unknown" bar and the same can be observed in the model score window with no feature scores being grayed out.
I do not know why I got that error and the same was observed with the skyline document I have shared with you through google drive.
Missing Library Intensity Dot Product:
I have not checked whether the target peptides of those decoy peptides without "library intensity dot products" have any matches. I will check it out let you know.
As far as I can remember, I have generated decoys soon after importing the spectral library without making any changes in any of the settings. So, I do not know how can a decoy peptide fails to sync with the target peptide, which is having spectrum information in the library.
Apart from this, I have also observed in my exported DIA results that many peptides with all of its MS/MS transitions identified with Q-value <0.01 and having idotp value > 0.7 and dotp value 0. How does this peptide is getting Q-value less than 0.01 and not having any dotp values?
I understand that dotp value was calculated when a product ion or MS2 transition dot product having a match with its respective library transition dot product. If that value found 0, that means no MS2 transitions are matching with respective library transitions or is there any cut-off such as intensity or mass error? Or Am I missing something here?
I have attached the DIA report file, where I have filtered such transitions having 0 dotp value and Q-value is less than 0.01. There are some other peptides with both idotp and dotp values are 0.
Thank You,
Chinmaya |
|
| |
| Nick Shulman responded: |
2019-12-30 16:08 |
What did you mean by "a few thousands of decoy peptides were not having any spectrum matches"?
I interpreted that to mean that you were saying that those decoy peptides did not have a library dot product-- hence my description of how to look for the "unknown" bar in the bar graph to find the peptides which are missing that feature. In the screenshot that you attached the "Library Dot Product" score is enabled, so you definitely do have that score available for all of the peptides in your document.
By the way, the data shown on the "Feature Scores" tab depends on which feature you have highlighted in the grid on the left. In your attached screenshot you are looking at the distribution of the "Intensity" scores. If you selected a different row in the grid, you would see the distribution of some other score.
I believe that the reason that it is possible to get a good q-value even if the library dot product is zero is because in your trained model the weighting on the "retention time difference" score is very high. In the Skyline document that you sent me, the weighting on the "Retention Time Difference" score was +714% and the weighting on the "Retention Time Difference Squared" was -621.7%. The net effect of these weightings is that the only thing that really matters in terms of getting a good q-value is how close the observed peak was to the retention time predicted by your iRT database.
With that model, it would definitely be possible to get a good q-value even if the library dot product was zero. Skyline could have chosen the peak based on the MS1 signal, even though the MS2 chromatograms in that region are completely flat. This would give you a library dot product of zero. However, the q-value would still be dominated by the retention time scores, so the library dot product would not matter much.
I do not understand how you could get a good peak where both the library dot product and the isotope dot product are zero. I see examples of that in your spreadsheet, but I do not see those in the Skyline document you sent me.
Is it possible that you might have manually adjusted the peak boundaries in Skyline? I believe that when you make changes to peak integration boundaries, the peak keeps its old q-value, even though that is a meaningless number if the peak location was chosen by a human.
-- Nick |
|
| |
| Chinmaya k responded: |
2019-12-31 00:09 |
Hi Nick,
Thank you for the clarification.
Yes, I got to see a few thousands of decoy peptides were not having any spectrum matches after checking on that "unknown" bar as I have mentioned in my previous response.
In my previous response, I have attached the screenshot of an error that occurred in the reintegration step despite the fact that all the features are enabled. The same I have observed in another DIA data search. I have shared the Skyline document through Google drive (OSCC_DIA_53W_Product_Ion_6_5_0.05Da.sky). Why mProphet model is failing to reintegrate? How to resolve the same?
Regarding transitions identified without MS/MS spectrum matches, Is it okay to accept such peptides/ transitions identified with Q-value <0.01 and not having single MS/MS spectrum matches?
Is there any acceptable range for "Retention Time Difference" or "Retention Time Difference Squared" or is it possible to set in Skyline?
The spreadsheet I have shared was not from the Skyline document I have sent you, So please do not compare. Sorry for the confusion I have caused.
However, I have such results where a peptide is identified with Q-value <0.01, idotp - 0 and dotp -0 without changing any peak boundaries.
I have attached a screenshot of peptide "ALQDQLVLVAAK" which is identified with charge state 2 (634.8823++) and 3(423.5906+++). Whereas, this peptide with charge state +3 has idotp, dotp values 0 and still qualifies with Q-value <0.01 though its corresponding decoy has idotp of 0.32 and dotp of 0.43 at RT 28.0 min unlike predicted RT of target peptide at 68.2 min. Here, I guess the "Retention Time Difference" is overriding all other features such as idotp and dotp values. I have shared the skyline document for the same through Google Drive (DIA_53W_Product_Ion_5_5_0.05Da.sky).
In both the case what I have mentioned above, the spectrum library, DIA raw file, Peptide and transition setting parameters were the same except changes in Product Ions picked for spectrum match. The product ion of 6 with minimum product ion of 5 has failed to reintegrate whereas product ion and minimum product ion of 5 & 5 respectively work perfectly fine.
The same raw file against the same spectrum library search gets reintegrated with Product ion and minimum product ion of 6 & 6 respectively (The Skyline document for the same has not sent through Google Drive).
Thank You,
Chinmaya |
|
| |
| Brendan MacLean responded: |
2019-12-31 07:06 |
That error message means there is insufficient separation between targets and decoys to continue with the mProphet method. That is, your targets are highly similar to your decoys. Can you click the Feature Scores tab and select some of the more important scores like Library intensity dot-product, Co-elution count, Shape (weighted), and Retention time difference? Take screenshots of what you see plotted for these scores and post them to this support thread.
Thanks. |
| |
| Brendan MacLean responded: |
2019-12-31 07:10 |
Also, based on what Nick got for his model, please uncheck Retention time difference squared. Because of the tendency (shown clearly in Nick's plot) for Retention time difference and Retention time difference squared to produce highly similar separation causing LDA to balance them in opposite directions with extreme values, we have removed Retention time difference squared from the next version of Skyline. We probably never should have added it, but in most cased (not yours) is has been relatively harmless. |
| |
| Nick Shulman responded: |
2019-12-31 12:12 |
Chinmaya,
Thank you for sending me your files.
When a peptide such as ALQDQLVLVAAK has more than one charge state, Skyline uses the maximum dot product value across all the charge states. Therefore, it's not unexpected that the chosen peak might have zero idotp and dotp values for one charge state. It's the other charge state's values that caused Skyline to choose that peak.
-- Nick |
| |
| Chinmaya k responded: |
2020-01-02 02:24 |
Hi Brendan,
Thank you for your support.
As per your suggestion, I have attached the screenshots of Feature Scores such as Library Intensity Dot-product, Co-elution count, Shape (weighted), and Retention time difference. These screenshots were taken from the Skyline document where mProphet failed to reintegrate.
The removal of the "Retention Time Difference Squared" feature score for reintegration has helped to train the mProphet model successfully. However, the RT prediction scores are getting disturbed and hence, the RT prediction calculation has to be performed again.
Does LDA use all the feature scores of mProphet?
Do all feature scores in mProphet has equal weight or Retention Time Difference, dotp and co-elution have more weight than other features?
Thank you,
Chinmaya |
|
| |
| Chinmaya k responded: |
2020-01-02 03:17 |
Hi Nick,
Thank for your support.
From your explanation, I believe that the "Co-elution count" feature score is playing a role in qualifying peptide charge states even though it does not have any idotp or dotp values.
Hence, I removed the "Co-elution count" and "Co-elution (weighted)" feature and reintegrated the peaks again. This has only reduced the number of such peptides from 20 to 17. Or else, are there any other features or parameters in Skyline causing this bug?
Thank you,
Chinmaya |
| |
|
|
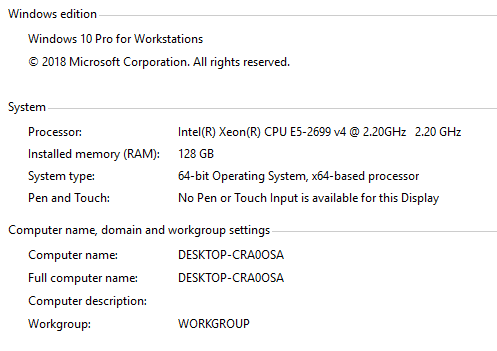 System_Configuration.PNG
System_Configuration.PNG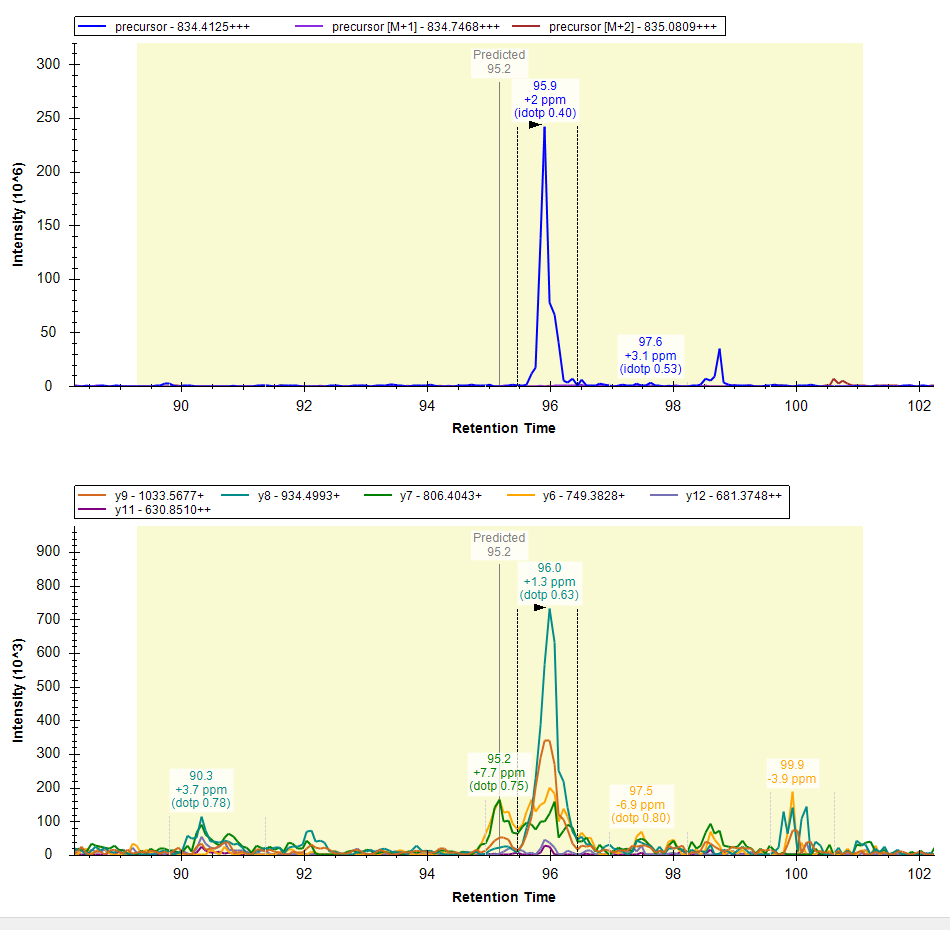 DIA_MS_MS2_Extracted_Chromatograms.PNG
DIA_MS_MS2_Extracted_Chromatograms.PNG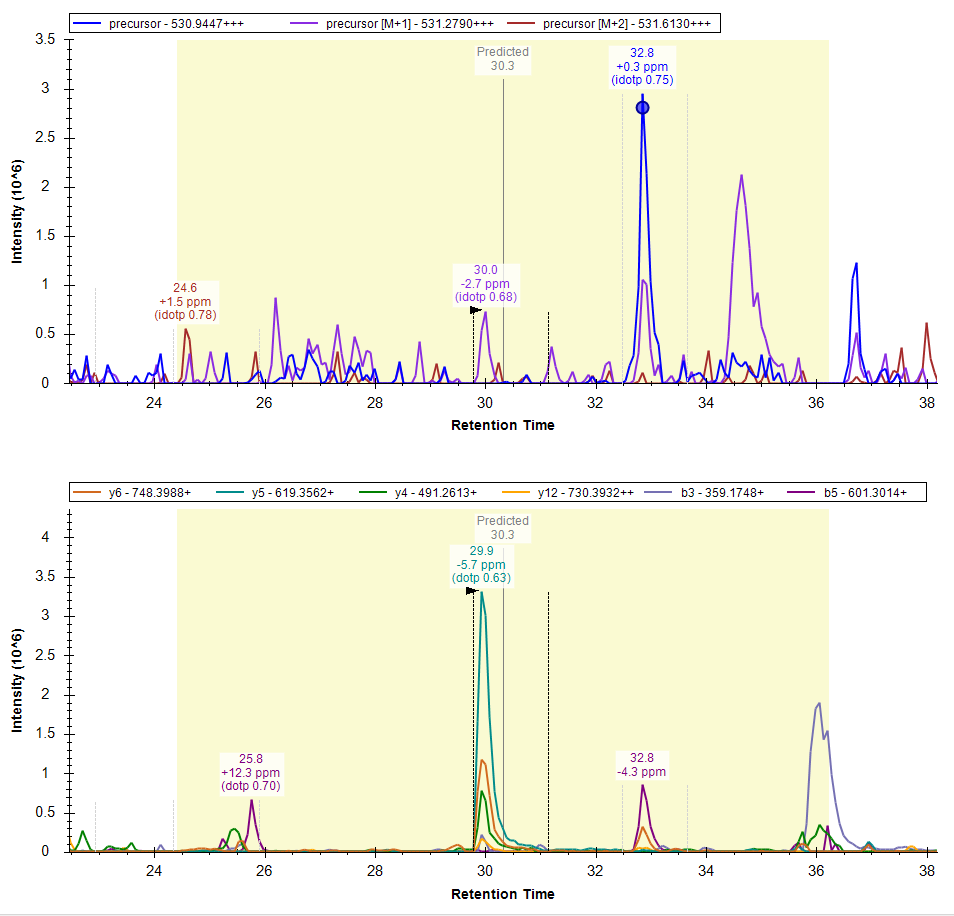 DIA_MS_MS2_Extracted_Chromatograms_02.PNG
DIA_MS_MS2_Extracted_Chromatograms_02.PNG Feature_Scores_Reintegration.PNG
Feature_Scores_Reintegration.PNG Reintegration_Error.PNG
Reintegration_Error.PNG OriginalModel.png
OriginalModel.png Reintegration_Error_123119.PNG
Reintegration_Error_123119.PNG Co-elution_count.PNG
Co-elution_count.PNG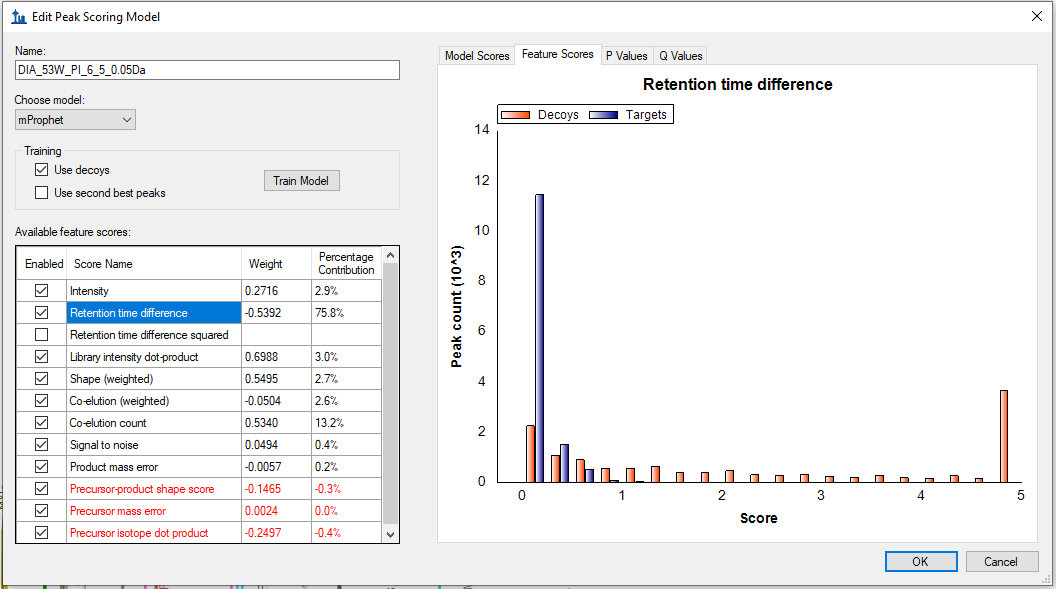 Retention_Time_Difference_Feature_Scores.PNG
Retention_Time_Difference_Feature_Scores.PNG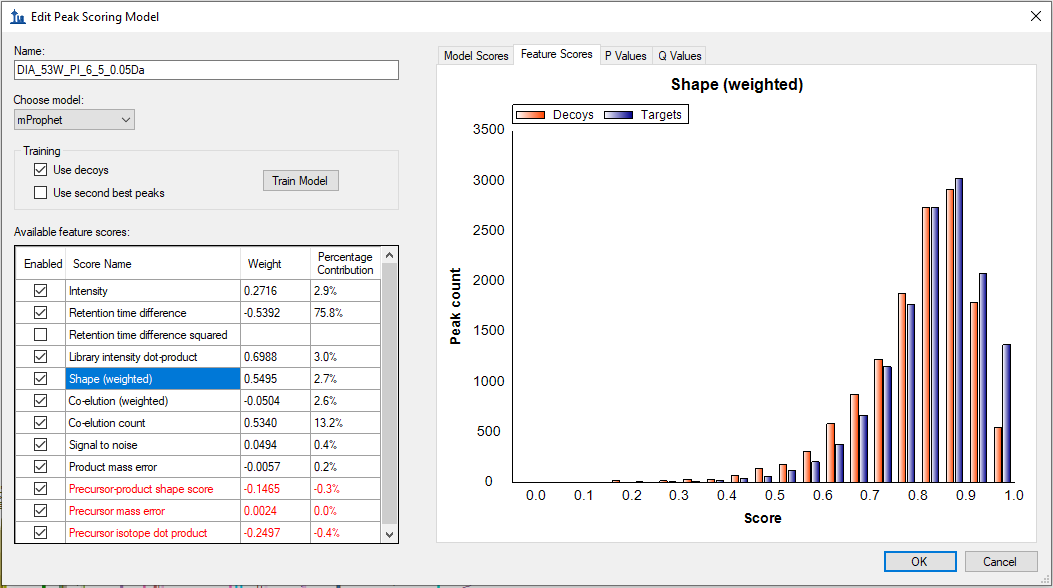 Shape(weighted)_Feature_Scores.PNG
Shape(weighted)_Feature_Scores.PNG