| Import QuanDirect (aka IS-PRM) data to Skyline | Markus | 2019-11-26 07:43 | |||||||||||||||||||||||
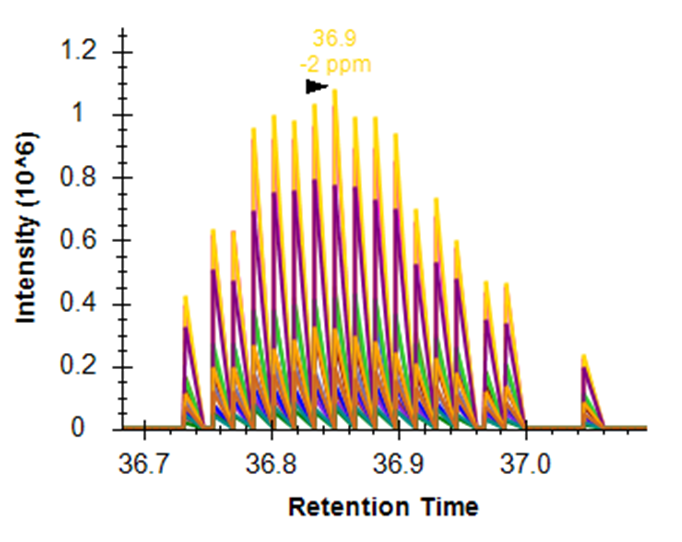
Hi Skyline team, we are experimenting with the QuanDirect (aka IS-PRM) method on the Orbitrap Lumos using heavy spike-in peptides and everything seemed to have worked fine so far. When importing the data into Skyline (daily 19.1.1.309) I run into the problem, that Skyline extracts all MS2 scans, i.e. also the ion-trap scans which are only targeted at the trigger y1 ion and which do not contain any other ion information. When combined with the MSMS Orbitrap scans that actually contain the fragment ions of interest this results in sawtooth-like peaks (see picture attached). How do I tell Skyline to only import/extract the hi-res Orbitrap PRM scans? Many thanks, |
|||||||||||||||||||||||||
| |||||||||||||||||||||||||
 quandirect.png
quandirect.png IT_and_OT_scans.png
IT_and_OT_scans.png