| Multiple precursors per peptide | sa825 | 2019-08-27 02:41 | |||||||||
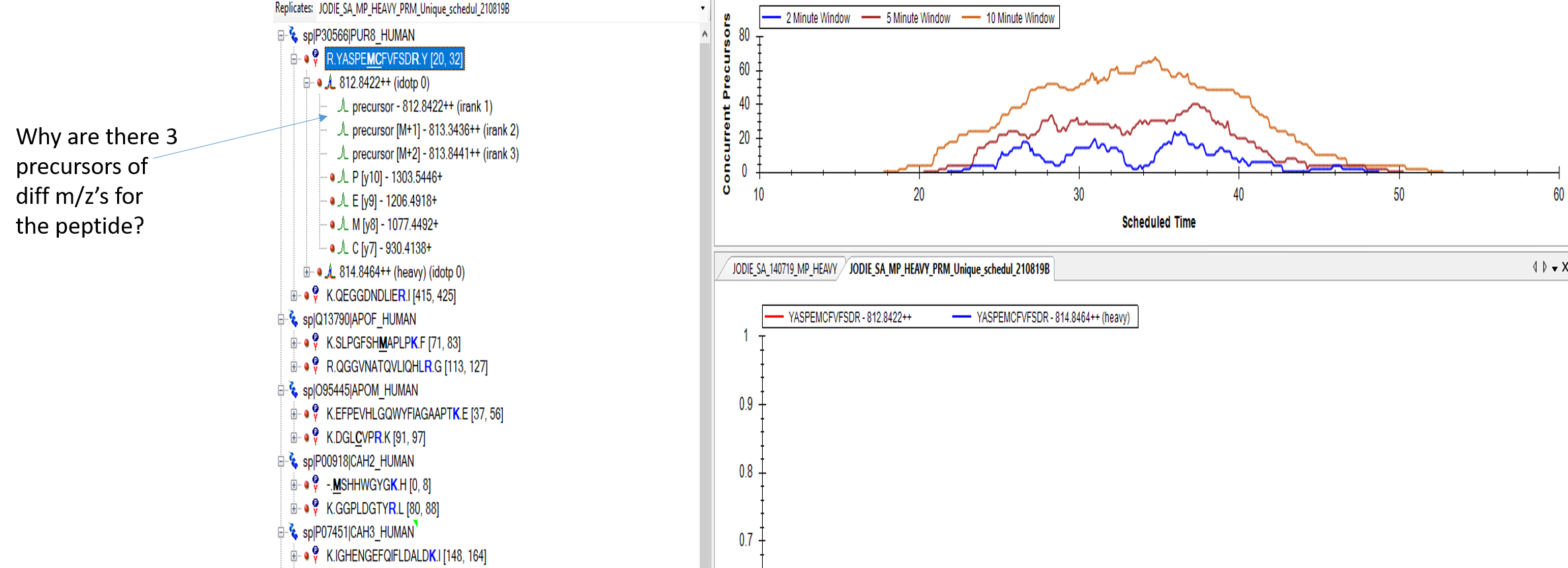
Good morning, I was wondering why there are 3 precursors of diff m/z’s for the peptide? And is it alright remove the ones with irank 2 & 3? The reason I am asking about this is because when I do a DDA run using the same peptides on the Thermo Q Exacitive, my peptide is detected at the expected retention time but when I do a targeted run (PRM), the Thermo is unable to detect my peptide. I fear its because the Thermo is looking for 3 precursors at the same retention time when it should only be looking for one? I have attached an image to show what I mean. Thanks, Shimon |
|||||||||||
| |||||||||||
 3 Precursors per peptide.png
3 Precursors per peptide.png