| |
| Nick Shulman responded: |
2019-08-11 21:29 |
This error can happen if you have two copies of the same peptide in your Targets tree, and the Explicit Retention Time that you have specified for those two copies of the peptide are different from each other.
This error happens because Skyline cannot decide what the predicted retention time for the peptide is.
There might be other ways for this error to happen. It would be helpful if you could send us your Skyline document so that we can see exactly what is going on.
In Skyline, you can use the menu item:
File > Share
to create a .zip file containing your Skyline document and supporting files.
If that .zip file is less than 50MB you can attach it to this support request.
Otherwise, you can upload it here:
https://skyline.ms/files.url
-- Nick |
| |
| Chinmaya k responded: |
2019-08-13 01:01 |
Dear Nick,
Thank you for your quick reply.
I was able to resolve that issue by removing peptide "AGEEGGSVGSGVFLIGR" from decoy list. However, apart from this small hurdle I am facing issue with overall DIA analysis with search speed, iRT prediction and reintegration with mProphet.
Here, I tried to do this DIA analysis in Skyline 19.1.0.193 using 2 windows workstation (Please find the attachments), working speed of Skyline starting from spectral library match, importing fasta file and further downstream analysis found extremely slow though.
In brief, I have used a spectra library (Created using Bibliospec) of respective samples acquired in 120min DDA run and analysed in PD 2.2 using Human proteome database from refseq. Pierce iRT peptides in each DDA samples were also spiked. Later, same samples with pierce iRT spike in DIA method of 12.5 Da X 56 window (ranging between 400-1100) were acquired and tried to match against the spectral library of the same.
I want know, why Skyline is not running smoothly in our 128 GB RAM windows workstation with such small dataset?
Why mProphet is not able to identify decoy features and reintegration was not working properly?
Are there any changes required either in our LC-MS/MS (DIA) method or DIA analysis parameters in Skyline to improve the search speed and reintegration?
As per your request, I have attached .zip file of the same analysis in the link you have shared with me earlier.
Let me know if you need more information.
Thank You,
Chinmaya |
|
| |
| Nick Shulman responded: |
2019-08-13 04:39 |
Thank you for sending us that file.
Skyline is spending an enormous amount of time updating the "Retention Times Peptide Comparison" window. If you close that window, I think you will find that Skyline becomes much faster.
I have not figured out yet why that window is so slow. You have 32000 peptides which is a reasonable number that I would not expect to cause performance problems for Skyline. I think the slowness might have something to do with missing values on that graph, but I am not sure.
Thank you for telling us about this performance problem. I will try to figure out what is going on and fix it.
-- Nick |
| |
| Nick Shulman responded: |
2019-08-13 12:23 |
Chinmaya,
The other thing that is making this document incredibly slow is the setting that you have at:
Settings > Peptide Settings > Digestion
You have "Enforce peptide uniqueness by" set to "Protein". You should change that to "None".
Every time any change is made to your Skyline document, Skyline goes through all of your peptides and figures out their uniqueness.
Meanwhile, Skyline is also querying the uniprot database in order to figure out the descriptions of the proteins in your Skyline document. Every time Skyline fetches the descriptions for a few more proteins Skyline needs to verify the uniqueness of your peptides as well as update the Retention Times Peptide Comparison window. Updating these two things take longer than fetching more protein descriptions, so it is almost impossible to click on anything in Skyline.
I will try to fix these issues.
-- Nick |
| |
| Chinmaya k responded: |
2019-08-14 03:01 |
Thank you nick for quick support.
Skyline is running quite smoothly, as I have changed "Enforce peptide uniqueness by" to "None". Inclusion of retention time predictor is not showing any issue with skyline speed.
Later, I re-analysed the same data with retention time prediction window of 5min. I also specified peptides with multiple protein matching to first matching protein while associating peptides from spectral library with background proteome (Human NCBI refseq). This helped me to train mProphet peak picking model with peptides having Q-value less than 0.01.
However, weight and % contribution for mProphet features such as "Retention time difference", "Retention time difference Squared" and "Library intensity dot-product" are not calculated. Why so? (Please find the attached mProphet_feature.csv file)
When it comes to peptide matching, it seems that peptides falling under the range of iRT prediction window were matched extensively. Only 450 transitions with RT more than 70min were matched out of 64765 transitions. How can I increase the match of peptides eluting in that range where iRT is not present?
From the exported result file, even though NCBI refseq proteome database was included as background database, I observed uniprot accession number for many proteins. Why it is so?
Finally, the current result section only gives transitions and peptides identified from the DIA data. How can I apply something like strict parsimony and calculate number of unique proteins identified from DIA analysis?
For information I have attached exported result file, mProphet feature and for more details I uploaded the .zip file in your file sharing folder (P_Y_CAM_Control_DIA_mProphet.sky.zip).
Let me know if you need more details.
Thank you,
Chinmaya
|
|
| |
| Chinmaya k responded: |
2019-08-14 04:12 |
Dear All,
I started analysing another set of DIA data with mProphet peak picking model. This time mProphet is able to calculate wight and % coverage for most of its features when trained. But, I am facing an error pop-up, when the same model was selected for reintegration.
Please find the screen shots trained mProphet and reintegration error.
Let me know if you need more info.
Thanks
Chinmaya
|
|
| |
| Nick Shulman responded: |
2019-08-14 12:50 |
Chinmaya,
You should use the menu item "Refine > Add Decoys" to add a new set of decoys to your document.
Some of the decoys in your document are still referring to the modification "Carboxymethyl (N-term)" which is no longer being used by any real peptides in your document. Because of this, I get an error when I try to open the .sky.zip that you uploaded.
(There is clearly a bug in terms of how Skyline decides that a modification is no longer being used in the document that I should fix)
In the Edit Peak Scoring Model dialog, you can use the "Feature Scores" tab to find all the peptides with a missing score. If you hover the mouse over the "unknown" bar on the bar graph, a binoculars button will appear below the graph, and if you click on the binoculars Skyline will populate the Find Results window with a list of the peptides that do not have that score.
If there are any peptides in your document that do not have a value for a particular score, then that score will not be available for model training. You should either delete those peptides from your document, or make it so that they have a score.
The "Library Dot Product" score is not available because Skyline requires at least 3 transitions to be willing to calculate Library Dot Product, and you have a couple of peptides (such as "LGSIDNCWPMLSIFFTEYK") that have fewer than 3 transitions.
You can tell Skyline to remove all peptides with fewer than X transitions by going to "Refine > Advanced > Min transitions per precursor".
The reason that "Retention time difference" is not available is that your iRT database only contains the iRT standards. There are not iRT values for the rest of the peptides in your document. That is, if you go to:
Settings > Peptide Settings > Prediction > Calculator Button > Edit Current
you will see that there are no rows in the lower ("Other iRT values:") grid.
-- Nick |
| |
| Chinmaya k responded: |
2019-08-18 02:34 |
Thank you Nick,
Yes, I have noticed carboxymethylation (N-term) and some other mod's being applied to the analysis by Skyline itself. I tried to remove them and do the DIA search, though peptides with such modifications found in the exported result file though.
Anyways, I am able to perform DIA analysis using mProphet model in Skyline. However, It would be great if the result file gives protein accession ID's from input proteome database not from uniprot.
Thank you,
Chinmaya |
| |
| Nick Shulman responded: |
2019-08-19 22:29 |
What do you mean by "It would be great if the result file gives protein accession ID's from input proteome database not from uniprot"? My understanding was that Skyline used the accession IDs to fetch info (gene name, protein name, etc) from uniprot.
Can you send us your FASTA file?
--Nick |
| |
| Chinmaya k responded: |
2019-08-20 00:03 |
In all of my report on DIA search, peptides corresponding to 1668 protein were identified. Where, 1280 proteins have accession ID from NCBI database (Used as background proteome) and 388 proteins have uniprot protein accession IDs. I have attached both the background proteome database used and DIA report.
For example, peptide "ANWYFLLAR" corresponding to protein "medium-chain specific acyl-CoA dehydrogenase, mitochondrial isoform a precursor" was identified with uniprot protein accession ID "P11310" in the exported DIA search report, when the same peptide corresponding to NCBI protein accession ID "NP_000007.1" can be seen in skyline (please find the screen shot).
Why Skyline is giving protein accession IDs from 2 databases when only one was used as background proteome database?
Thank you,
Chinmaya |
|
| |
| Chinmaya k responded: |
2019-08-20 00:14 |
I am not able to attach any documents to my previous response. An error is being showed by this portal.
500: Unexpected server error
org.postgresql.util.PSQLException: ERROR: duplicate key value violates unique constraint "uq_documents_parent_documentname"
Detail: Key (parent, documentname)=(864f789d-a532-1037-bd53-e465a393a22b, Screenshot_ANWYFLLAR_NP_000007.PNG) already exists.
DialectSQL=INSERT INTO core.documents
(_ts, createdby, created, modifiedby, modified, owner, container, parent, documentname, documentsize, documenttype, document)
VALUES (?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?, ?)
RETURNING rowid
|
| |
| Nick Shulman responded: |
2019-08-20 12:45 |
I do not know why you are getting that error when you try to attach files to this support request.
If you would like, you can upload your files here:
https://skyline.ms/files.url
I will also send you an email message in case it's easier for your to send files over email.
-- Nick |
| |
|
|
 Windows8_Workstation.PNG
Windows8_Workstation.PNG Windows10_workstation.PNG
Windows10_workstation.PNG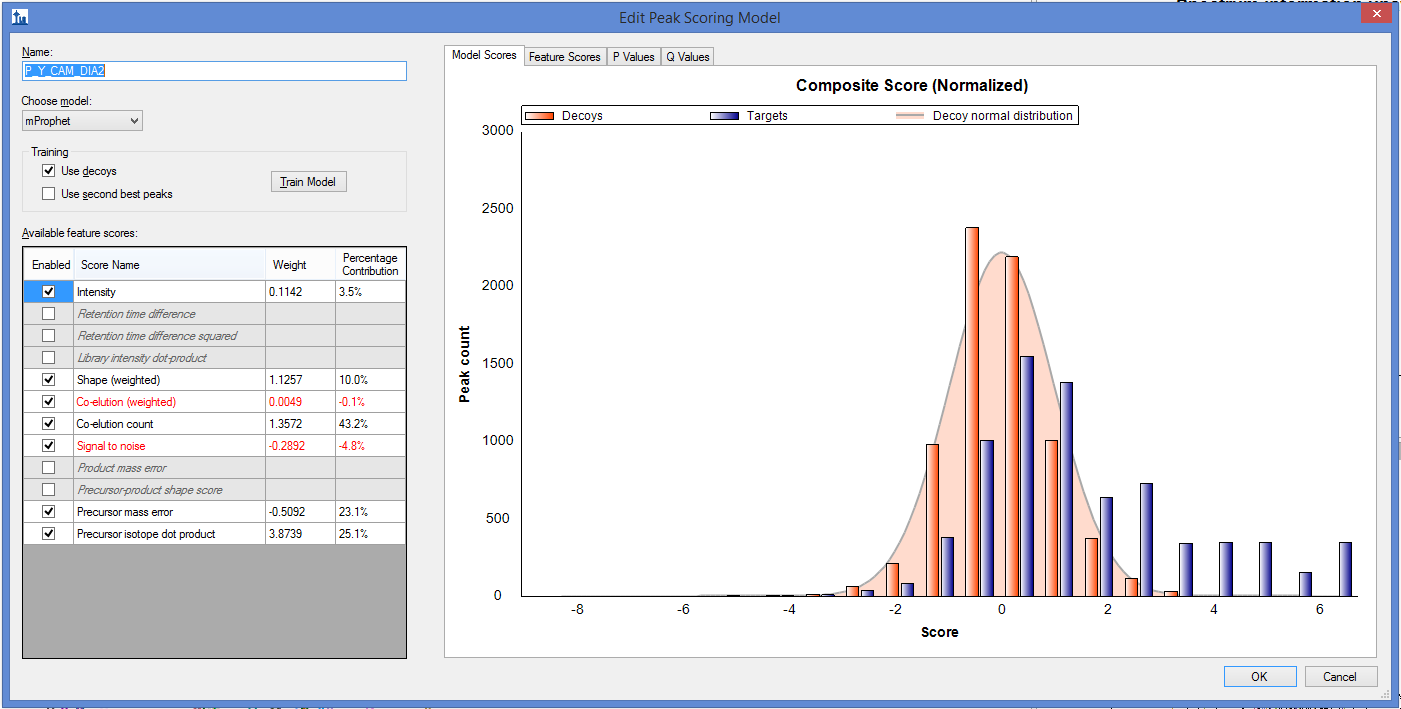 mProphet_trained_model.PNG
mProphet_trained_model.PNG Reintegration_error_mProphet_model.PNG
Reintegration_error_mProphet_model.PNG Screenshot_ANWYFLLAR_NP_000007.PNG
Screenshot_ANWYFLLAR_NP_000007.PNG