Hi Sunberg,
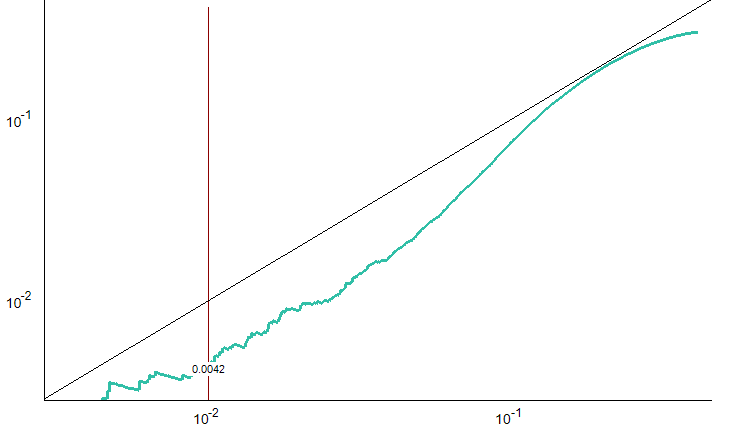
You appear to be using Edit > Refine > Compare Peak Scoring, which is intended for use with datasets which have been manually curated with all integrated peaks in the document considered "True-Positives". Then you compare some other way of scoring and picking the peaks with this ground truth. The "Observed False-Positive Rate" comes from comparing the alternate scoring method with the document as it is currently integrated. Anything that would pick something other than what is in the document is considered a false-positive.
So, I guess, the question remains: where do the two cases you are comparing come from:
- The existing integration boundaries
- The alternately scored integration boundaries
For, instance, if #1 is simply the default peak picking in Skyline and #2 is a trained mProphet model, then your Q-Q plot is not valid and you are not looking at useful information, because every "false-positive" is simply a case when the mProphet model would change the integration from the default peak picking.
Hope this helps to clarify. The Peak Scoring Comparison UI was really intended for researchers working on implementing these scoring models, like the ones that contributed to the Navarro, Nature BioTech 2016 paper:
https://www.ncbi.nlm.nih.gov/pubmed/27701404
Thanks for clarifying the source of your Q-Q plots.
--Brendan
 qq.PNG
qq.PNG decoy01.PNG
decoy01.PNG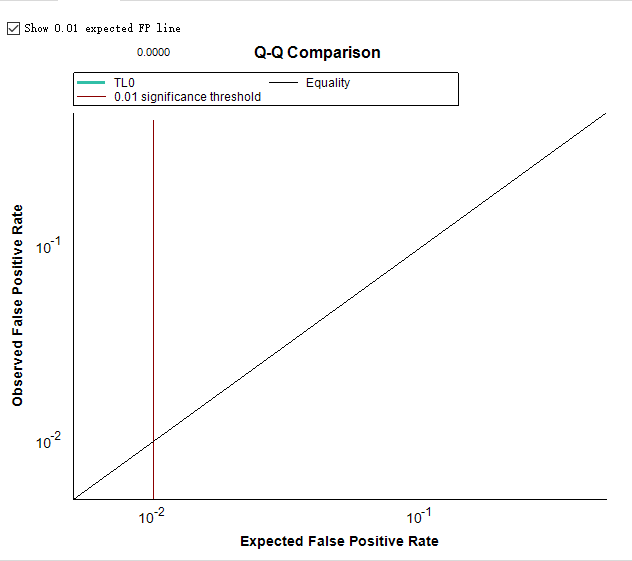 qq.PNG
qq.PNG