| Nick Shulman responded: |
2019-03-10 20:12 |
You should send us your Skyline document. We will figure out what is going on.
In Skyline, you can use the menu item:
File > Share > (complete)
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms and iRT database.
You can upload that .zip file here:
https://skyline.ms/files.url
I imagine that the problem is that some peptides do not have entries in the .iRT database, but it might be that something weird is going on.
-- Nick |
| |
| Brendan MacLean responded: |
2019-03-10 21:36 |
That vertical line of points on the left of you retention time regression plot is indicating a set of peptides in your document which Skyline cannot find in your iRT library. We can't give those the value zero. So, we need to choose a negative value which we usually try to make left of the rest of the distribution by some amount. So, I don't totally understand why that line appears to connect with the rest of the distribution, but these are definitely the peptides without a matching entry in the iRT database.
Are you able to find what you think are these entries in your iRT database? As Nick points out, it might be that they are there but are not matching for some reason.
Do you actually have a .irtdb file, or are your iRTs inside your .blib file by virtue of how you built your library? If you click the calculator button shown in your Snapshot4.PNG and click Edit Current, the form that appears should show you the path to the file that contains your iRTs as well as a list of iRTs which you could select and copy into Excel to search.
Definitely interesting that they are so consistently N-terminal modified peptides which seem to match your library spectra. If your iRTs come from your .blib file, then there probably is some kind of bug in the way the iRT values get written to the .blib file during library creation.
Giving us your files as Nick suggests should help. Thanks for the report with the screenshots.
--Brendan |
| |
| Nick Shulman responded: |
2019-03-10 22:24 |
It looks like all of your peptides with missing retention time differences are decoy peptides.
When Skyline generates decoy peptides by shuffling the sequence or whatever, Skyline remembers what original peptide the decoy was generated from. For things like retention time difference, Skyline knows that it is not going to find the shuffled sequence in your iRT database, so Skyline looks up the original sequence instead, and pretends that that's the predicted retention time for the shuffled sequence as well.
One of your peptides that is missing a predicted retention time is "PKSDGQ[-17.026549]QEDLDK". If you look in the .sky file with a text editor, you can see that that peptide was generated by shuffling the original peptide "Q[-17.026549]LEDGDQPDSKK". That original peptide is no longer in your Skyline document, and does not have an entry in your iRT database.
I think you should generate decoys again, reimport results, and then try training your model again.
-- Nick |
| |
| Brendan MacLean responded: |
2019-03-12 11:24 |
Nick is right about regenerating your decoys and reimporting. Did that work?
I would also recommend you make sure the Library dot-product score is enabled. That should be one of your strongest scores.
You can get it enabled using Edit > Refine > Advanced (Refine > Advanced in Skyline-daily) and set the "Min transitions per precursor" field to 6 (3 MS1 and 3 MS/MS).
This should get rid of all of the peptides you currently have with just 2 fragment ions, which is not enough to generate a library dot-product (minimum 3).
There is also a setting you can use for this in Transition Settings > Library where you can set "minimum product ions", which you may want to set as high as 6 as it has been set for some published studies. Especially when you have your desired (or maximum) fragment ions to 10.
Hope this helps.
--Brendan |
| |
| anatoly.urisman responded: |
2019-03-12 18:46 |
Brendan and Nick, thank you for the replies.
Unfortunately, I am still getting the same behavior. For some reason, these particular peptides are not getting added to the iRT table.
I have followed your advice as follows (a couple of times now):
1. Removed decoy peptides
2. Set "Min transitions per precursor" to 7
3. Removed empty peptides and proteins
4. Added decoy peptides (shuffled)
5. Reimported results
6. Added results to iRT table (replaced existing) - see new regression plot (attached snapshot)
I don't see anything unusual about these peptides when I examine them manually (e.g. selected peptide in the snapshot). Virtually all have reasonable peaks correctly identified right next to the blue ID lines.
Should I upload my raw files for you to try?
Thanks again for your help!
-Anatoly |
|
| |
| Nick Shulman responded: |
2019-03-12 21:51 |
Can you send us your .sky.zip again?
It sounds like there might have been two sets of peptides that are not present in the iRT database, and the decoys were distracting us from the other set.
-- Nick |
| |
| Brendan MacLean responded: |
2019-03-13 07:39 |
Note also that it is not usually appropriate to train you iRT values to your analysis data set. So, once you do this training, you can't really rely on you mProphet model statistics, since you are essentially saying you expect to find your peptides where you found them in your targets, but you are not training your decoys in the same way. You are still saying you expect to find your decoys at the times where you found their source peptides, which is not fair if you trained off the targets in your samples.
You need to do a separate experiment for training iRTs than you the one you use them in.
But, yes, send us files to look at. We clearly do not yet understand what is going on with your modifications. Ideally, you would send us the file exactly is it was when you took the screenshot.
--Brendan |
| |
| anatoly.urisman responded: |
2019-03-13 10:19 |
Brendan, your point about iRT is well taken - I agree 100%. This is a brand new iRT calculator that I took straight from this dataset for convenience. I will update it with data from a different run.
The latest .sky.zip file is on the share. It is unchanged from the time the last snapshot (6) was taken. I also think that there is something weird going on with the modifications. Is there something about the mass decimal precision of these particular modifications in my library?
Thanks!
-Anatoly |
| |
| Nick Shulman responded: |
2019-03-13 11:02 |
There seems to be a bug in Skyline 4.2 where peptides that have modifications of negative mass (e.g. "M[-89.02992]") can never be found in an iRT database.
This is fixed in Skyline-Daily. We did not know about this bug, but other things changed so that the erroneous code never gets used.
I would recommend that you start using Skyline-Daily with this dataset, because I'm not sure whether or when a fix for this will make it into Skyline 4.2.
If you open up this document in Skyline-Daily, it works great, and you will be able to train an mProphet model that uses the retention time difference.
Sorry about this bug, and thanks for letting us know about it!
-- Nick |
| |
| anatoly.urisman responded: |
2019-03-13 11:23 |
Thank you very much for working through this issue with me. Always appreciate your help and the fantastic job you are doing for the proteomics community!
-Anatoly |
| |
 Snapshot1.PNG
Snapshot1.PNG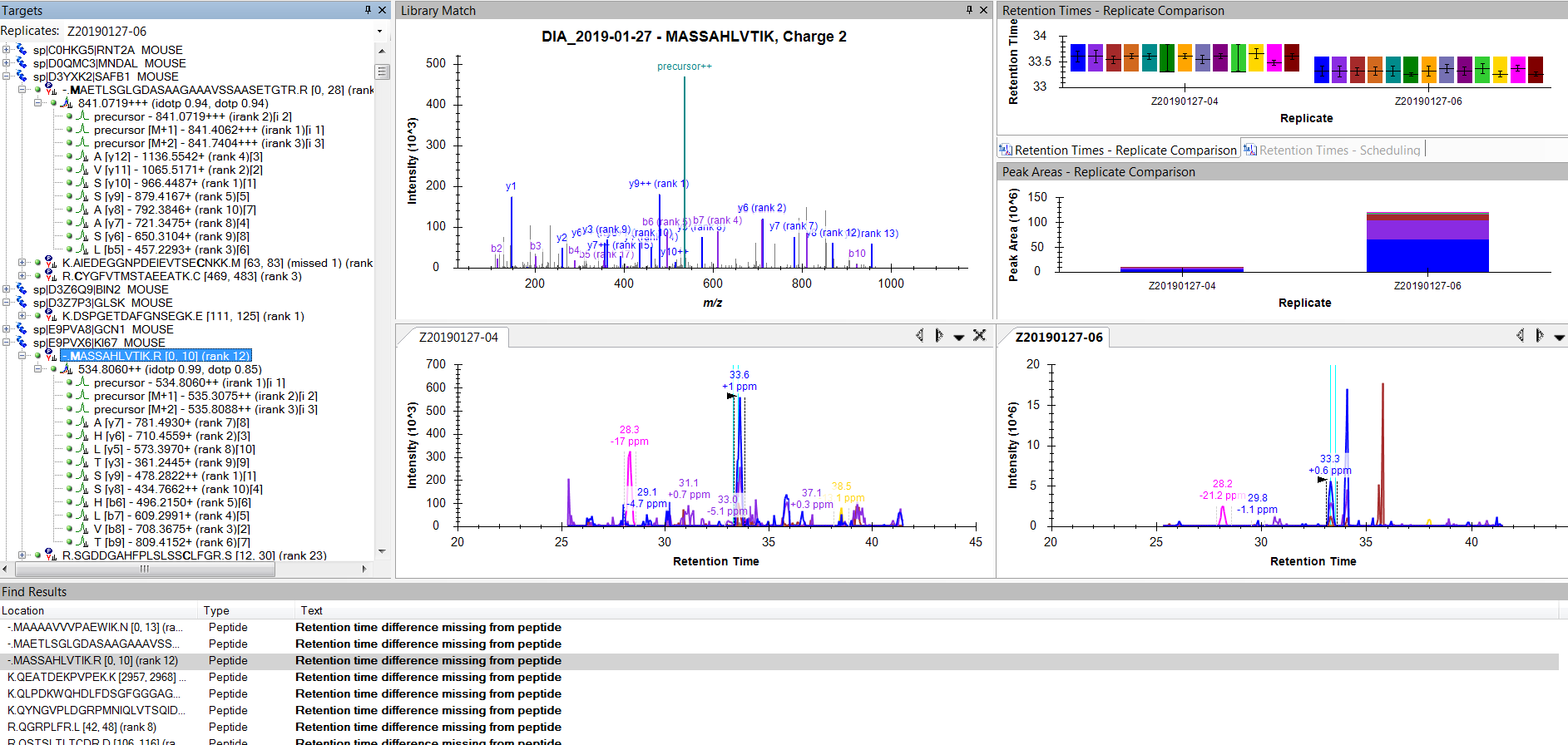 Snapshot2.PNG
Snapshot2.PNG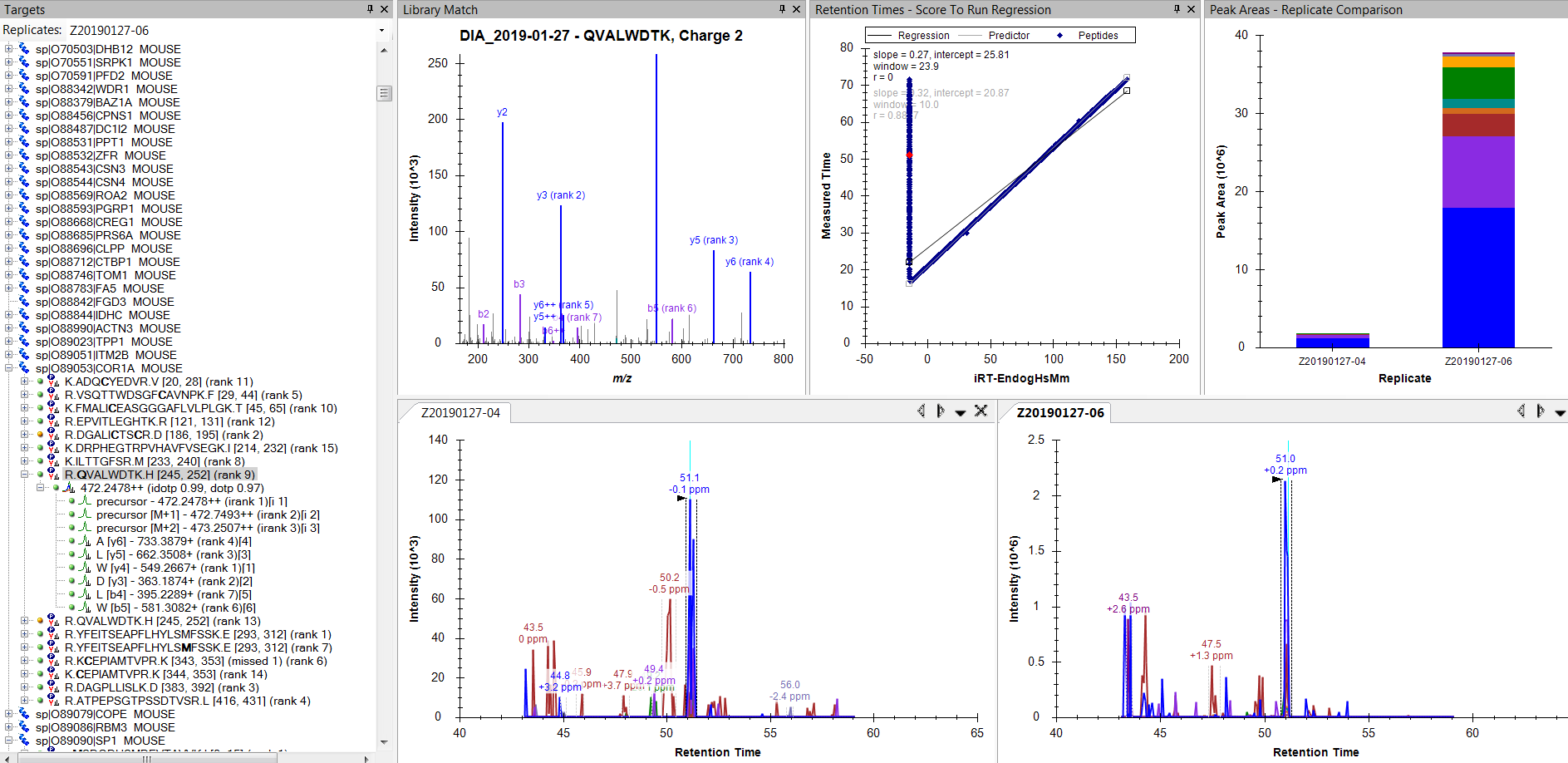 Snapshot3.PNG
Snapshot3.PNG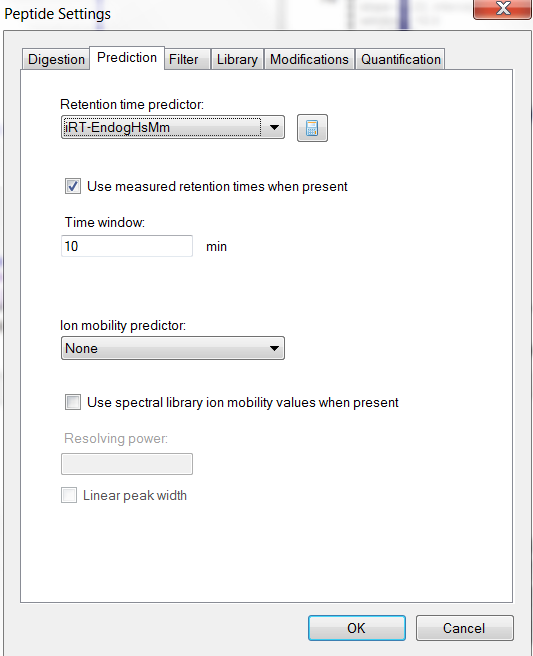 Snapshot4.PNG
Snapshot4.PNG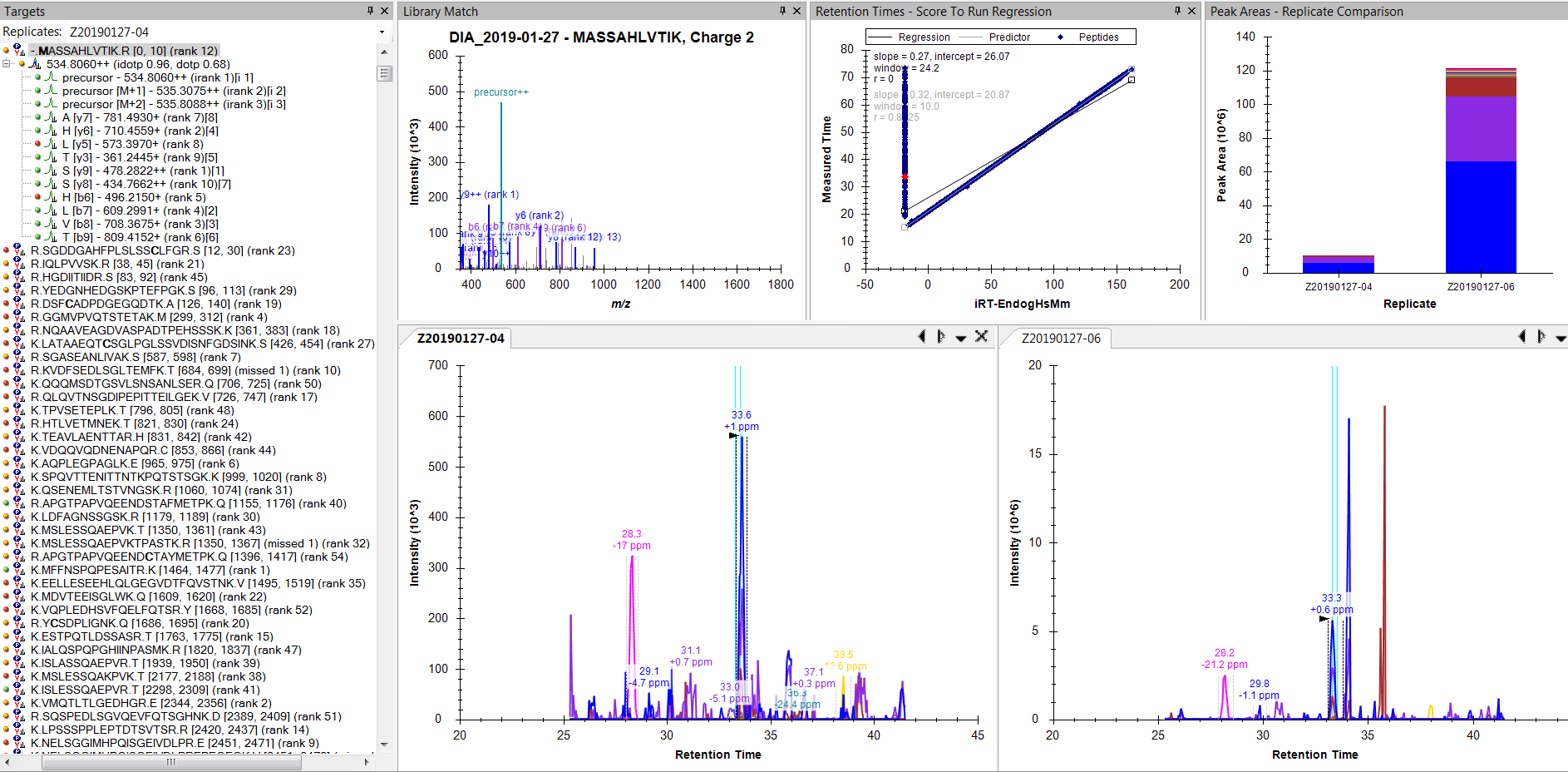 Snapshot6.PNG
Snapshot6.PNG