| |
| Nick Shulman responded: |
2019-02-14 03:44 |
Is your .dat file from Mascot search results? Skyline knows how to read Mascot search results. You should not try to convert them to anything else.
Here is the list of all of the search result formats that Skyline knows how to read:
https://skyline.ms/wiki/home/software/BiblioSpec/page.view?name=BlibBuild
On Page 4 of the full scan filtering tutorial, when you press the "Add Files" button, you will be able to select files with many different filename extensions, including ".dat".
When you are working through the steps of the tutorial, you should use the files that the tutorial tells you to, which can be found inside of the .zip file that you download at the beginning of the tutorial.
When you want to analyze your own data, you should have no problem getting Skyline to read your Mascot .dat files.
Let us know if you have any trouble.
-- Nick |
| |
| t j f m voermans responded: |
2019-02-14 04:01 |
Dear Nick,
The .dat files are from the Waters Xevo G2 QTOF. When I add the files I get the message: "ERROR: Unknown imput file type"
I wanted to attach the files that were created by the Waters Xevo G2 QTOF to maybe make things more clear, but there seems to be a server error. So I can only send one.
Thank you for helping!
Tim |
|
| |
| Nick Shulman responded: |
2019-02-14 04:23 |
I believe Waters raw files are actually entire directories, and all of the files inside of the directory are important.
Skyline knows how to read Waters .raw directories. The way that you tell Skyline to extract chromatograms from these .raw directories to use the menu item:
File > Import > Results
However, before you tell Skyline to extract chromatograms, you need to tell Skyline which peptides or molecules you are interested in. If you have peptide search results, then the MS1 full scan filtering tutorial will show you how to start from those search results and analyze your data.
If you have a list of peptides that you want to analyze, then you can tell Skyline about them by using the menu item:
Edit > Insert > Peptides
Skyline does not have the ability to just open a mass spec file and show you the spectra, etc. If you just want to browse the spectra in a Waters file, then you can use a program called "SeeMS.exe" which is part of ProteoWizard. You can download ProteoWizard here:
http://proteowizard.sourceforge.net/download.html
Waters has probably given you a better tool for looking at mass spec data than what SeeMS.exe is, but SeeMS.exe has the ability to look at multiple different formats of mass spec raw files.
Do you have a list of peptides you want to analyze?
-- Nick |
| |
| t j f m voermans responded: |
2019-02-14 05:39 |
Thank you for the link to the software.
I am looking at the effect of various parameters playing a role during tryptic digestion of BSA. Because I couldn't load the data in Skyline I looked up the mass of every peak I could find in the mass spectrum and made skyline do a trypsin digestion simulation on the sequence for BSA. Then I checked by hand if the value of the peptides predicted by skyline were the same as an m/z value found in the MS spectrum. However, instead of doing everything by hand. Skyline should be able to match the fragments found in the mass spectra with the predicted peptide fragments.
So the only list of peptides that I have is those created by the trypsin digestion simulation done by skyline. |
| |
| Nick Shulman responded: |
2019-02-14 06:06 |
It sounds like you successfully told Skyline about the peptides that you are interested in. The next step is to tell Skyline how to extract chromatograms for the data.
Did you try using the menu item:
File > Import > Results
and did Skyline give you some sort of error message?
Skyline might give you the message "To extract chromatograms, full scan settings must be enabled". If that happens, then you need to go to:
Settings > Transition Settings > Full Scan
and make changes to the "MS1 Filtering Isotope Peaks Included" or "MS/MS filtering acquisition method" section.
Can you send us a screenshot of what you are seeing in Skyline? I am not sure where you are getting stuck.
-- Nick |
| |
| t j f m voermans responded: |
2019-02-14 07:06 |
I did get the message "To extract chromatograms, full scan settings must be enabled" and then I choose for MS1 Filtering:
- Isotope peaks included: Percent
- Precursor mass analyzer: TOF
- Min % of base peak: 20%
- Resolving power: 30.000
- Isotope labeling enrichment: Default
and then 'Ok'
After that, I imported the raw data file and it did seem to work (I saw all the mass spectra being imported in different colours). But then I get 'Chromatogram information unavailable' (see pictures) |
|
| |
| Nick Shulman responded: |
2019-02-14 07:29 |
You are almost there.
The next thing that you need to do is tell Skyline that you want to extract precursors (from the MS1 scans).
To do that, go to:
Settings > Transition Settings > Filter
and under "Ion Types" change it to "p" for "precursor".
If you wanted your chromatograms extracted from MS1 and MS2, you might make that "y, p" or "y, b, p", but, since you only want the precursors, then you can just make that "p".
After you change the ion types, Skyline will change the set of transitions that are in your Targets tree.
If you expand one of those peptides in the Targets tree, you will see at one level the precursors for the different charge states. If you expand the precursors, you will see that Skyline has given you a few precursor transitions ("M" for the monoisotopic precursor, "M+1" for the isotope that is one Dalton heavier, etc).
If you have already imported results, then the chromatograms might show up immediately (you told Skyline to extract MS1, but since the transitions did not exist in the Targets tree, Skyline did not show them to you).
Otherwise, you can do:
File > Import > Results
-- Nick |
| |
| t j f m voermans responded: |
2019-02-15 02:23 |
That's it! I got the results
Thank you so much. With this information, I am sure I will be able to figure out the other great functions of Skyline with the help of the tutorials.
I am sure I am able the find the information somewhere, but is there a way to increase the specificity? Because some peptides were found during the whole run and have a deviation of 14,5 ppm. So there is only a small chance, if any, that these peaks are from the peptide itself (see picture).
Thank you again for taking the time to explain everything in great detail. |
|
| |
| Nick Shulman responded: |
2019-02-16 21:39 |
With data like this, where there are only MS1 chromatograms, I believe the only thing that Skyline pays attention to is the whether the peak areas of the M, M+1, M+2 etc match the expected isotope distribution based on the chemical formula of the peptide.
It is possible to get Skyline to pay attention to the mass error, but that involves training an mProphet model, and you probably do not want to do that, but this is the tutorial:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=tutorial_peak_picking
I am not sure what type of experiment you ran. If this was a DDA experiment, where after each MS1 scan the top N peaks were selected for further fragmentation and analysis in an MS2 scan, then you should run a peptide search program such as Sequest, Mascot, XTandem, or one of the many other peptide search programs.
Skyline does not know how to do any sort of DDA peptide search at the moment, but Skyline does know how to read a lot of different peptide search result formats.
This is the whole list of peptide search results that Skyline knows how to read:
https://skyline.ms/wiki/home/software/BiblioSpec/page.view?name=BlibBuild
-- Nick |
| |
|
|
 Home screen.png
Home screen.png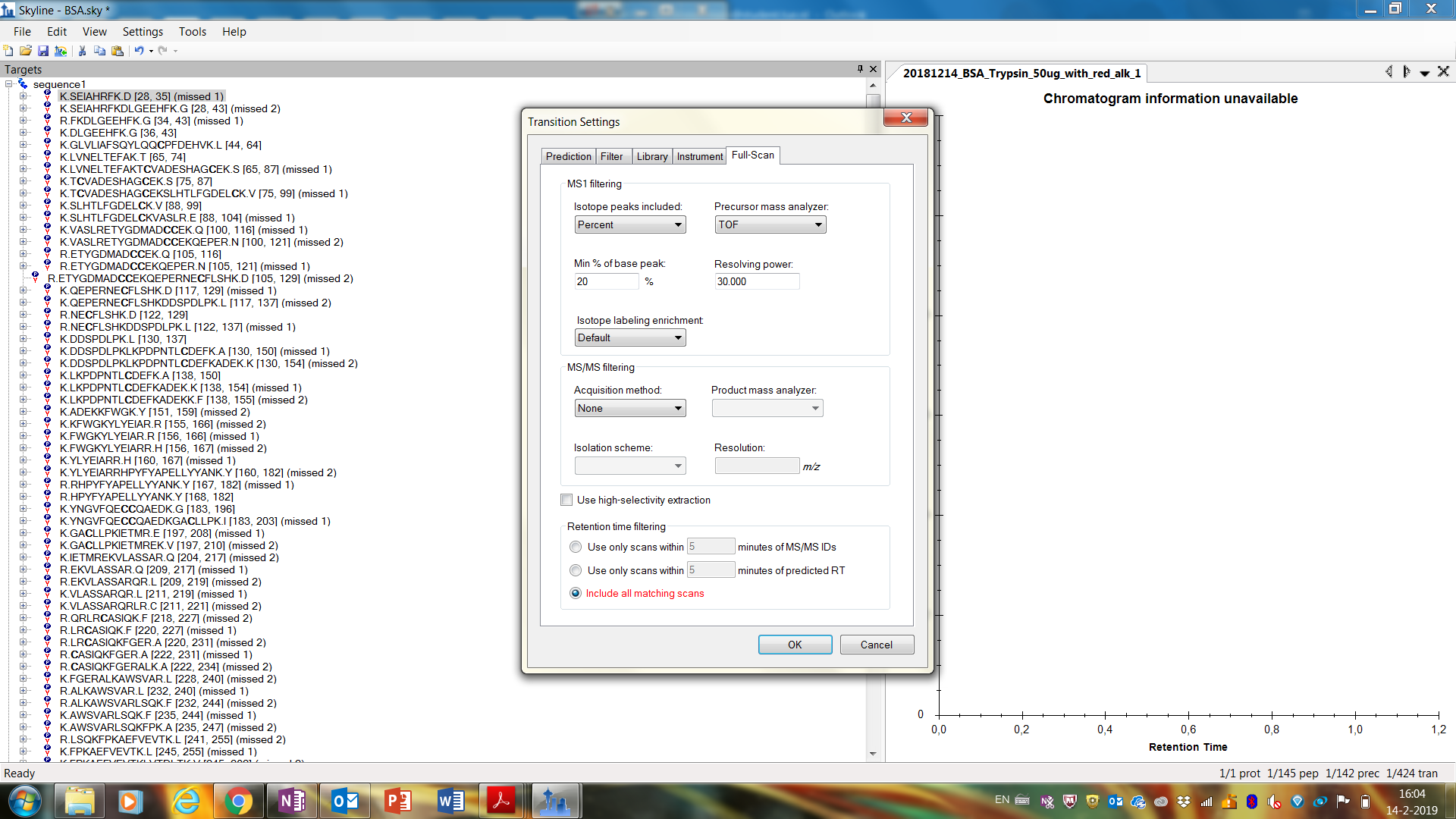 Transition settings.png
Transition settings.png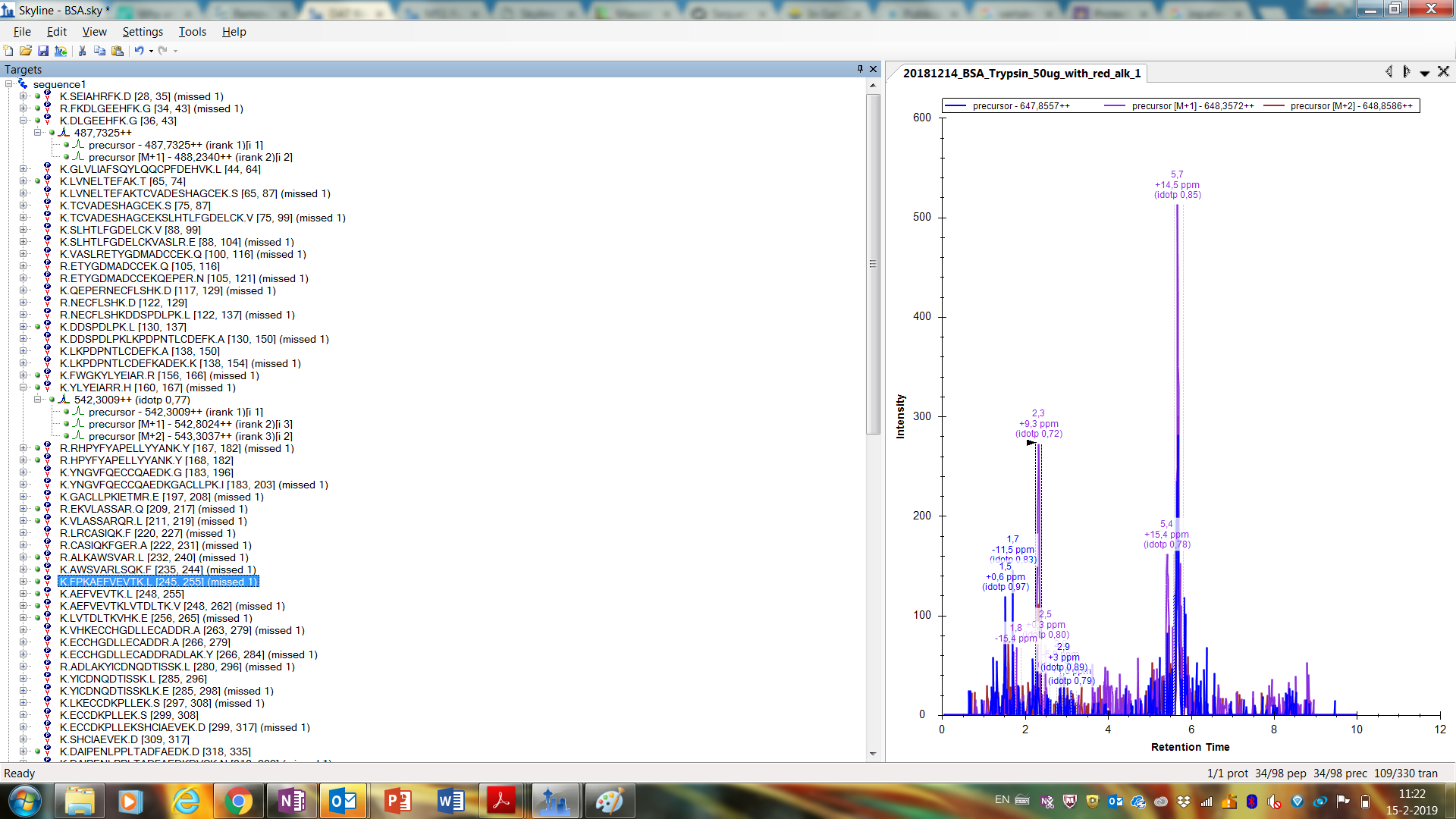 14,5 deviation of peptide.png
14,5 deviation of peptide.png