Hi Michael,
I think you are getting close. If you truly want only 6 fragment transitions, then you are choosing the wrong option "From filtered ion charges and types plus filtered product ions" on the Transition Settings - Library tab. This will give you the union of your 6 transitions and everything else in your heuristic transition filter.
You want to choose either "From filtered ion charges and types" (which will ignore your other filter settings) or "From filtered product ions" which will apply your entire filter and only use library peaks which meet your filter criteria. The MacCoss lab often uses an "ion 3" to "last ion" filter and the "From filtered product ions" option.
It is highly unusual to choose the option you have chosen, and I have considered removing it entirely because of its potential to cause confusion.
I am not clear, however, on how you got to where you have chromatograms extracted from only a subset of the transitions in your Targets list. It seems like maybe you had different targets when you last re-imported your data files.
Anyway, you are close. Try one of the two options I have suggested to prove to yourself that you can get a set of targets with 6 product ion transitions matching your library and properly including neutral loss ions. Then and only then, re-import your data files, and you should be set to do your analysis.
Thanks for the new screenshots. Good luck with your research.
--Brendan
 sc2.PNG
sc2.PNG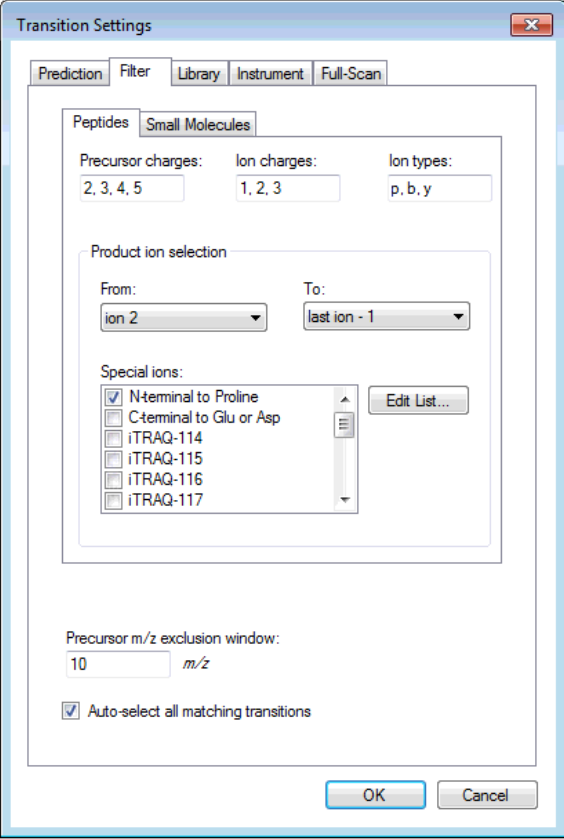 sc3.PNG
sc3.PNG sc1.PNG
sc1.PNG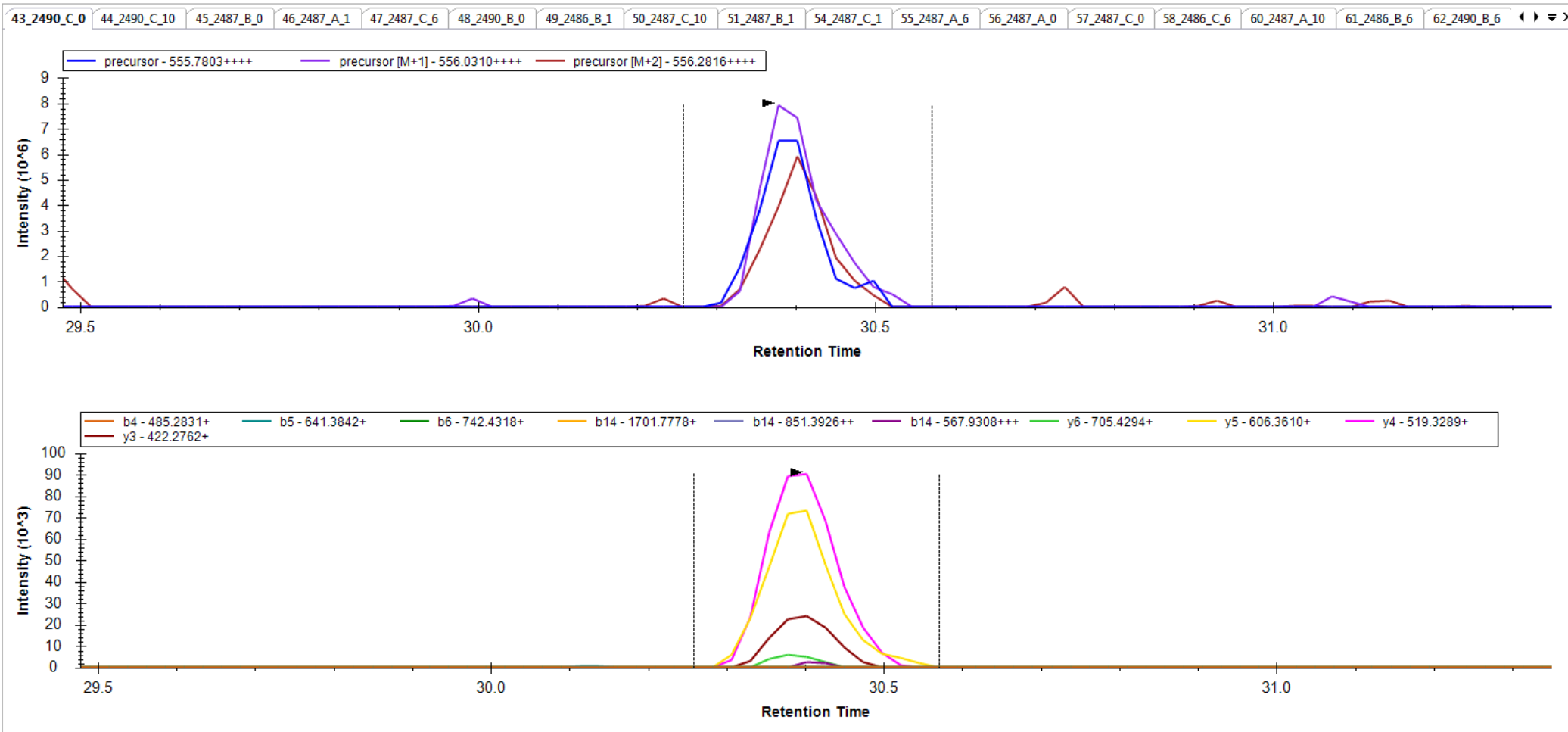 sc4.PNG
sc4.PNG sc5.PNG
sc5.PNG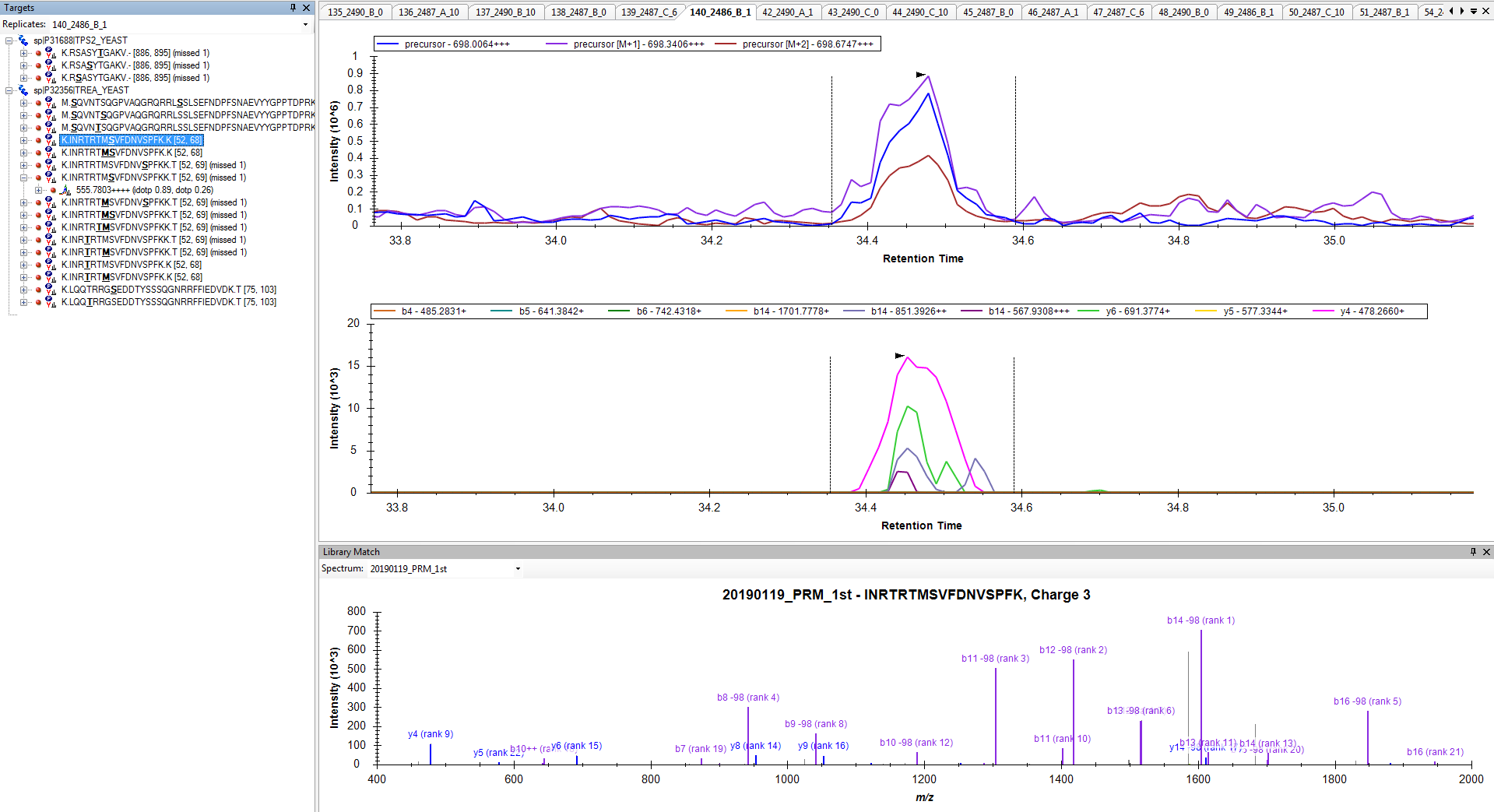 sc6.PNG
sc6.PNG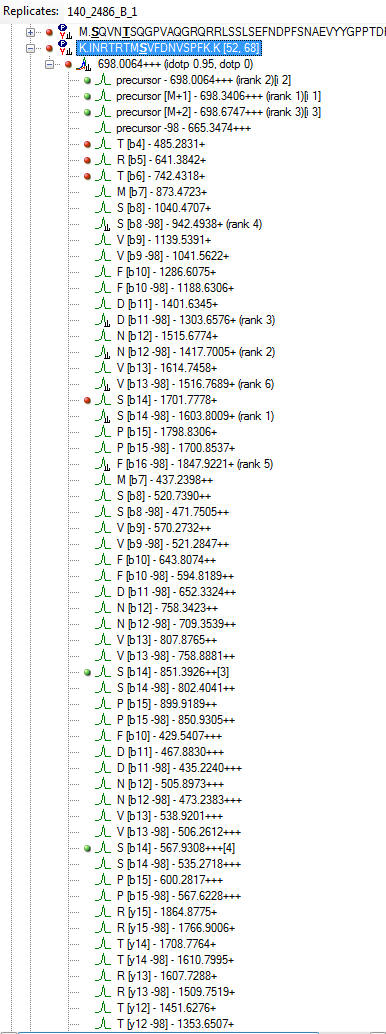 sc7.PNG
sc7.PNG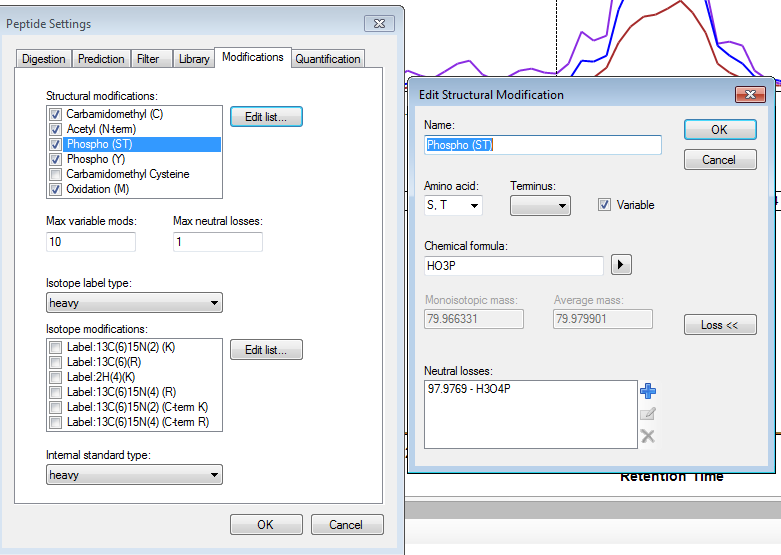 sc8.PNG
sc8.PNG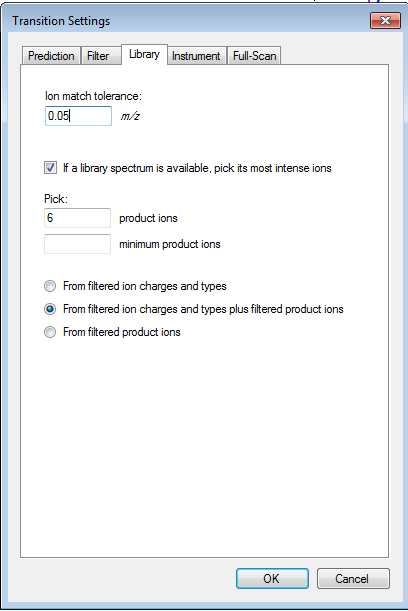 sc9.PNG
sc9.PNG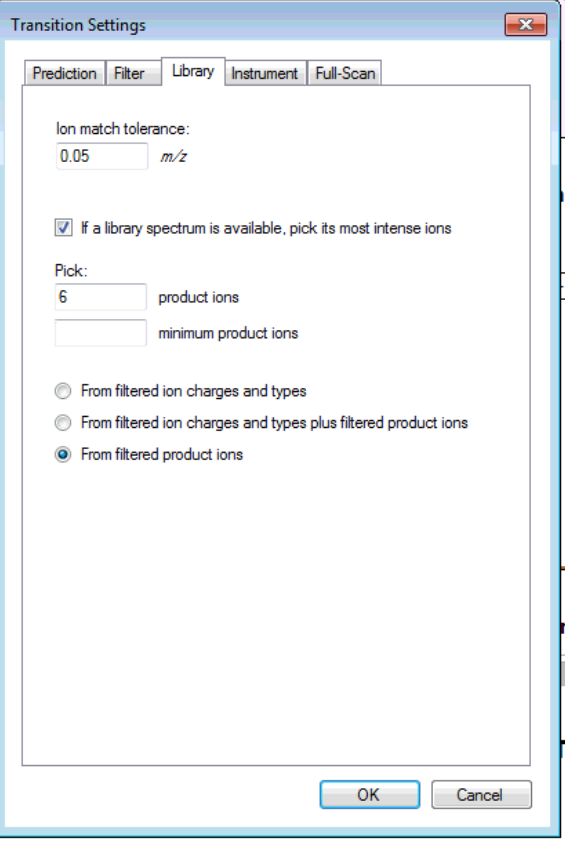 sc10.PNG
sc10.PNG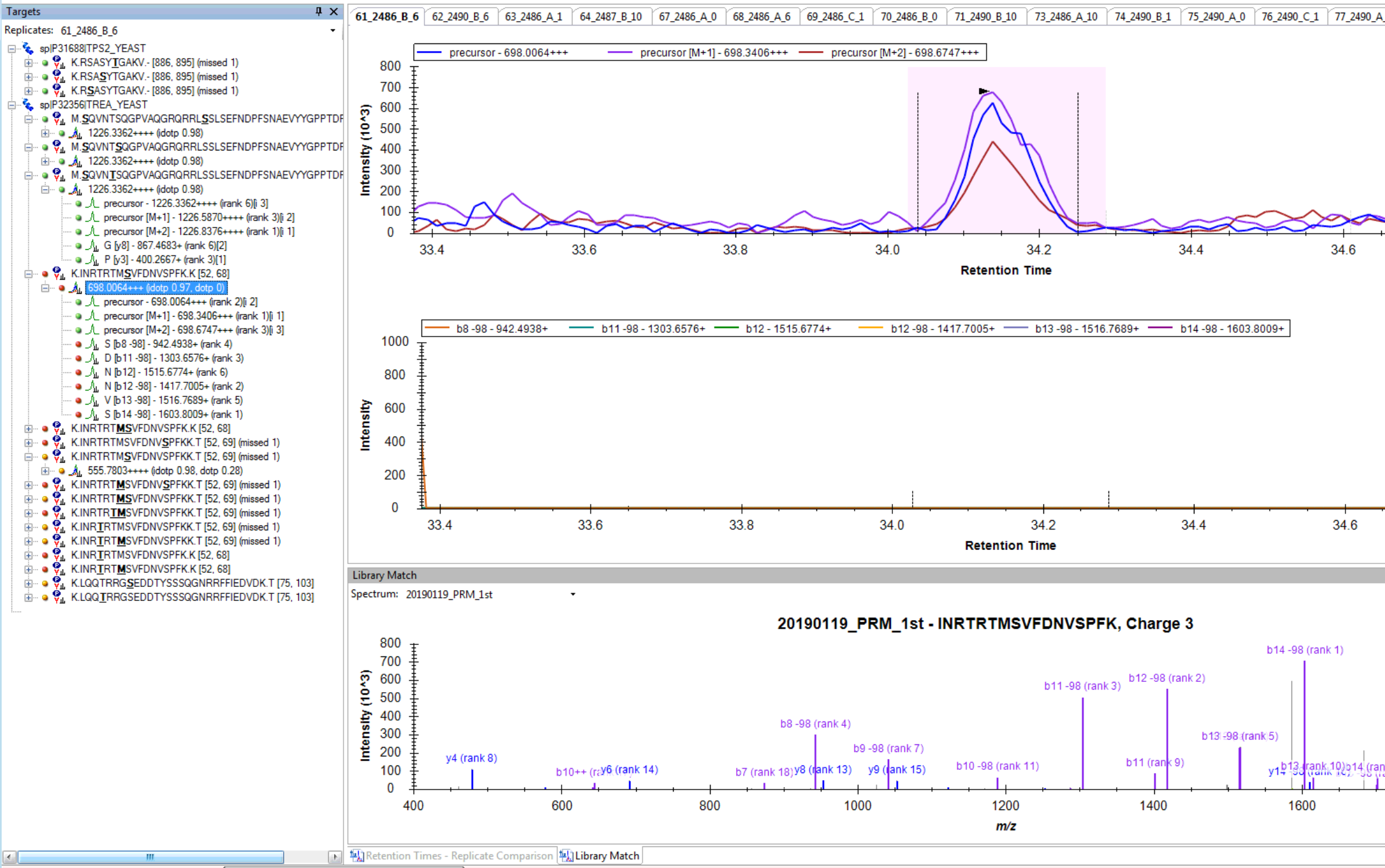 sc11.PNG
sc11.PNG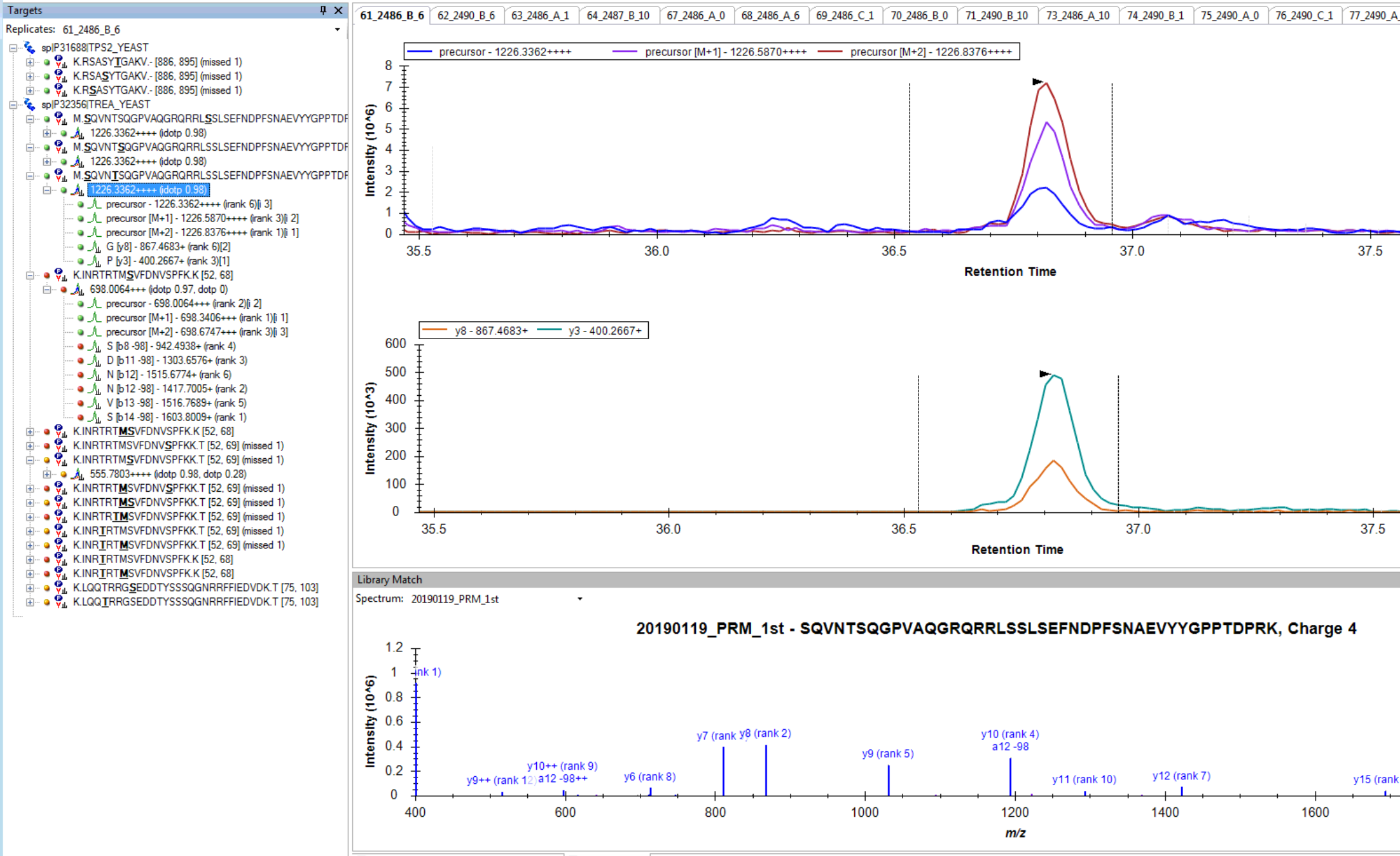 sc12.PNG
sc12.PNG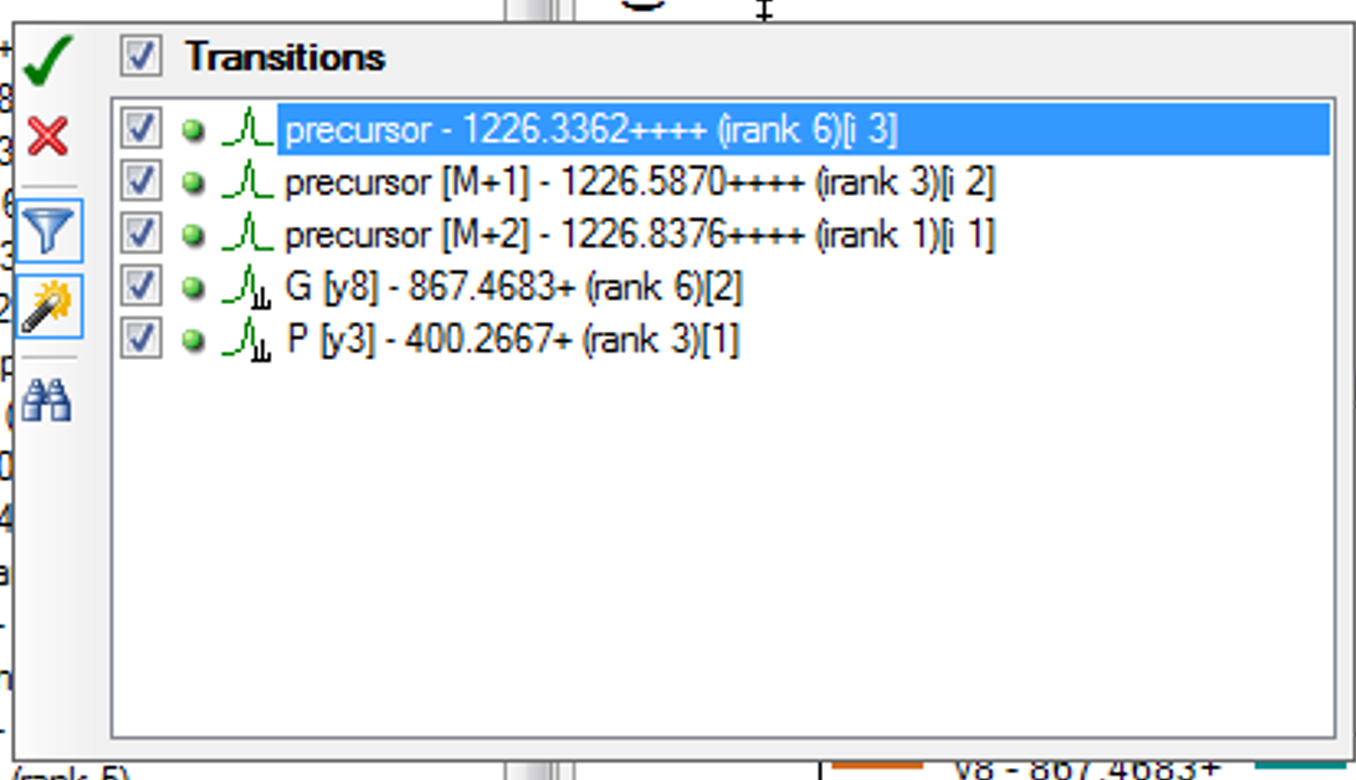 sc13.png
sc13.png sc14.PNG
sc14.PNG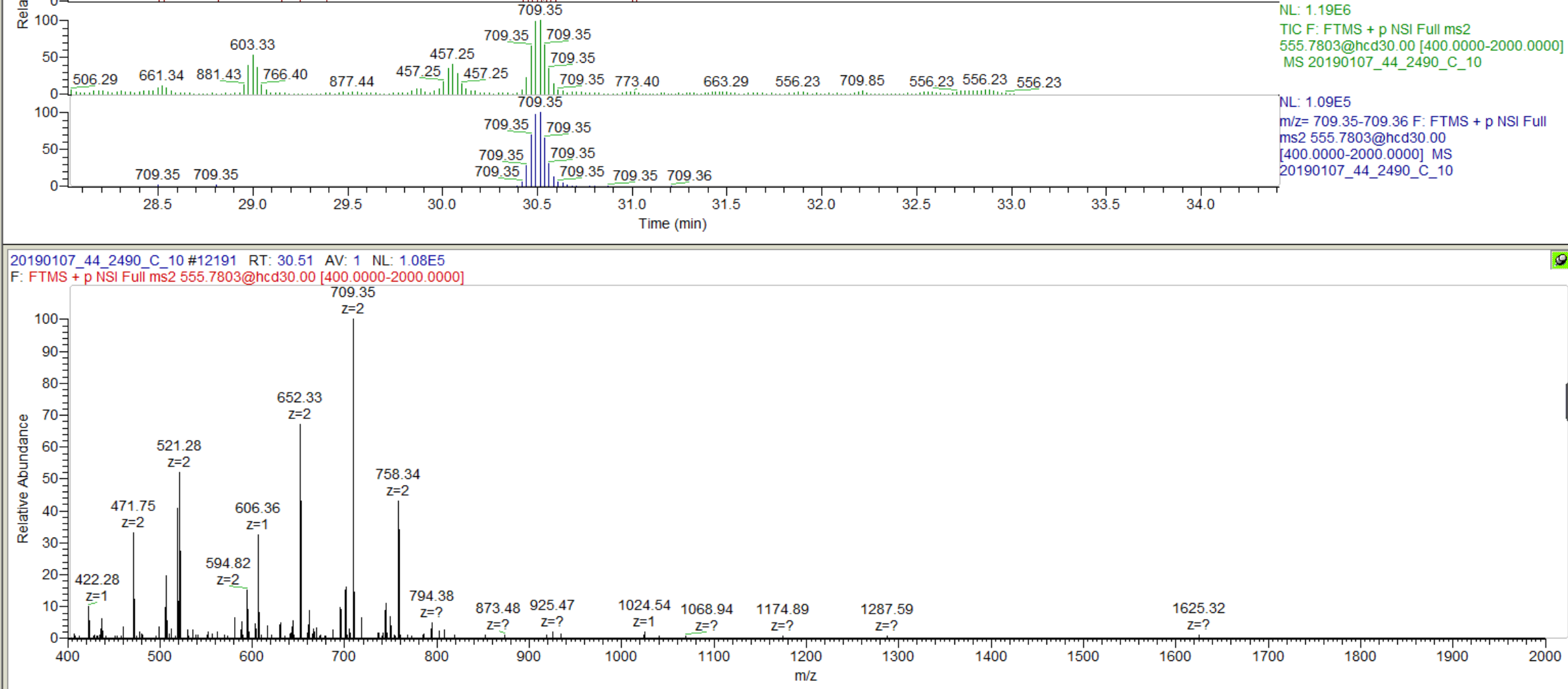 sc15.PNG
sc15.PNG