Hi all,
When I import with MS1 filtering (Isotope peaks included:Count, Precursor mass analyzer: Orbitrap, Peaks:3, Resolving power: 60,000@200m/z) and MS/MS filtering (Acquisition method: Targeted, Product mass analyzer: Orbitrap, Resolving power: 60,000@200m/z) I get split peaks on some chromatograms (Untitled1.jpg). If i remove MS/MS filtering and set it to none and reimport my data the full peak is restored (Untitled2.jpg). If I switch back to the original settings (MS1 and MS/MS filtering) and reimport, my peak is again split (Untitled3.jpg). I am using skyline 4.2.0.180305. Split peaks seem to be rare. I have a large document and most peaks are fine. I think that the split peaks occur on singly charged peptides with m/z >1000 but I would need to look into that. Any advice would be appreciated. The reason I tried turning off MS/MS filtering was that I found an old issue that restored a peak under this scenario (https://skyline.ms/issues/home/issues/details.view?issueId=294&_docid=issue%3A294). Am I doing something wrong or is there a bug somewhere in there? Thanks for any advice.
Best,
Michael
|
| |
| Nick Shulman responded: |
2018-11-30 06:45 |
Can you send us your Skyline document?
In Skyline, you can use the menu item:
File > Share > (complete)
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file is less than 50MB, you can attach it to this support request.
Otherwise, you can upload it here:
https://skyline.ms/files.url
It would also be helpful if you could send us one of your .raw files.
-- Nick |
| |
| Brendan MacLean responded: |
2018-11-30 08:33 |
Hi Michael,
This looks to me like the consequence of specifying your MS/MS Acquisition method as "Targeted" instead of DIA (or in the current Skyline-daily as DDA). If you use "Targeted", then the MS/MS spectra are assumed to have been acquired systematically to measure your targets of interest, now mostly referred to as PRM (the full-scan equivalent of SRM/MRM). In this case, we decided to only extract MS1 chromatograms over the same range as the MS/MS spectra cover, since it is now common for PRM to be measured in a scheduled mode where the MS/MS are acquired only over a scheduled range of retention time.
This makes perfect sense for PRM, but it doesn't make sense at all for DDA, which we really never expected to produce useful MS/MS chromatograms, certainly not for quantification, because the MS/MS spectra for each target are not acquired systematically.
The current available ways around this are:
1. In Skyline 4.2, you would need to use the MS/MS Acquisition method "DIA" with an Isolation scheme based on "Results", which means the MS/MS spectrum isolation target (and optionally window) is used. If you just use the default "Results" isolation scheme, then anything that falls within the 1 to 2 m/z isolation window that is common for DDA will have its fragment ions extracted. If want a more limited window, like 0.05 m/z, you need to define that yourself.
2. In Skyline-daily, you can choose the new Acquisition method "DDA", which will not limit your MS1 extraction based on the MS/MS and it will automatically set all your fragment ion targets to Quantitative = False.
If you think I have missed the mark on this explanation, then definitely get Nick the files he has requested.
Thanks for posting to the support board with screenshots.
--Brendan |
| |
| Michael Cundell responded: |
2018-12-03 02:33 |
Hi Both,
Thank you for your help. That all makes sense now. We are running DDA and PRM within a single method. DDA was acquired across the whole retention time, whilst PRM was done with scheduling. I have multiple scheduled PRM windows spanning the majority of the retention time across which I wanted to extract additional MS1 chromatograms from the DDA data. Exactly as you suggested Brendan, the split peaks lie within RT windows not covered by any PRM window and where DDA MS2 fragment ions matching my skyline document were detected thereafter which the MS1 precursor data is extracted (but not before).
I also hit another problem with this sort of DDA/PRM method in the past ( https://skyline.ms/announcements/home/support/thread.view?entityId=2f06739f-9769-1036-8cc6-e465a393e3d4&_docid=thread%3A2f06739f-9769-1036-8cc6-e465a393e3d4). Presumably such combined methods create a bit of a headache. I will try your workarounds and see how I get on. Thanks once again!
Best,
Michael |
| |
| Michael Cundell responded: |
2018-12-03 06:22 |
Actually scrap that.
Apologies, I should have stated exactly what I am doing upfront. I spiked a sample with 30 heavy labelled peptides to get a rough quantitation of their endgenous counterparts from DDA MS1 chromatogram data (I also needed the discovery data). I also spiked a further 3-4 peptides for quantitation with scheduled PRM. Samples were run on an Orbitrap Fusion Lumos with a method comprising alternating 1sec DDA then scheduled PRM and so on across the full retention time window.
On closer inspection the split peaks I observe occur in the 30 heavy labelled peptide DDA MS1 chromatogram data both within and outside of any PRM windows scheduled for the different 3-4 PRM precursors. Some of the 30 DDA peptides have MS2 data that demarks the split peak boundaries, others also have similar MS2 data without their peak being split. Unfortunately, I am not allowed to send this data through to you to take a look at which is quite annoying.
Those peaks that are split are restored using DIA import method you suggested Brendan. Thank you. However, I think I might separate out my skyline document. The PRM data can be extracted using Targeted MS/MS filtering with no MS1 filtering (albeit I have to deal with the spikes in the PRM data from DDA scans as described in https://skyline.ms/announcements/home/support/thread.view?entityId=2f06739f-9769-1036-8cc6-e465a393e3d4&_docid=thread%3A2f06739f-9769-1036-8cc6-e465a393e3d4). The DDA part can reside in a separate document that can be extracted with "DDA" using skyline daily which as you describe will do exactly as I hoped for that section of the data.
Thanks!!
Best,
Michael |
| |
| Brendan MacLean responded: |
2018-12-03 08:19 |
Yike. Mixed PRM and DDA. That is something we haven't seen or considered yet.
In hindsight, it sounds like it would have been best to somehow put your PRM peptides on an exclusion list for DDA to avoid having the DDA algorithm pick them for separate isolation.
Otherwise, I think you could achieve this with the mock-DIA I described and you seem to have tested. You would just want to set the Quantitative property on the fragment transitions yourself, which you can do in the Document Grid.
Or you can use two separate documents, as you describe. Thanks for the feedback and letting us know what you are having to face in getting this working.
--Brendan |
| |
|
|
 Untitled-1.jpg
Untitled-1.jpg Untitled-2.jpg
Untitled-2.jpg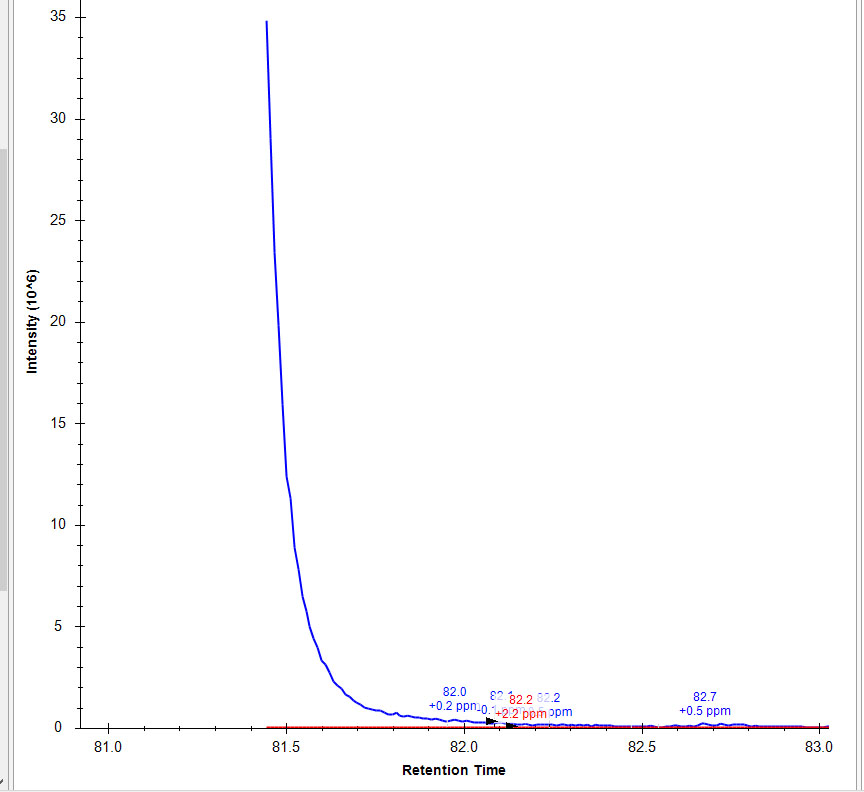 Untitled-3.jpg
Untitled-3.jpg