Dear skyline team,
I have a weird issue when trying to import a custom assay library.
I have set up a skyline document with lots of flexibility in terms of transitions to pick which is probably not a great approach but was supposed to do for now.
When I now import my assay library, skyline will generate the spectral library with peak m/z's as given in the assay library.
The document that is created though, will include transitions which are further off the spectral library than should be allowed (tol. set to 0.001).
I have a peptide for example, that has a y10 transition in the assay library.
This y10 transition is also in the spectral library that gets created.
In the actual transition list of the document, I get instead y11 - 71.1.
While this transition is close by, there is really no reason skyline should pick this, as it is too far off and skyline even sees it as y10 in the spectral library.
I use skyline 4.1.0.18169
|
| |
| Nick Shulman responded: |
2018-09-17 09:17 |
Can you send us your Skyline document?
In Skyline, you can use the menu item:
File > Share > (complete)
to create a .zip file containing your Skyline document and supporting files, including spectral libraries.
If that .zip file is less than 50MB, you can attach it to this support request. Otherwise, you can upload it here:
https://skyline.ms/files.url
-- Nick |
| |
| jfoe responded: |
2018-09-18 00:49 |
|
|
| |
| Nick Shulman responded: |
2018-09-18 10:42 |
The reason that Skyline is giving you that y11-71.1 transition is that you have a bunch of Losses defined that have been set to "Include Always" (the default is "Include if Matches Library".
You can control whether transition losses are included always by going to:
Settings > Peptide Settings > Modifications > Structural Modifications > Edit List...
And then you can edit one of your neutral loss modifications (e.g. "Deamidation (N-term) + Loss").
Then, when you are editing the modification, the "Edit Neutral Loss" button might be hard to find, so I'm attaching a picture of it.
There, you can change "Include loss by default".
The original value for these "Include loss by default" was, I think, "Matching Library", and I think you changed that to "Always".
For this reason, Skyline had to give you the y11-71.1 transition.
That transition, by the way, has the following losses on it:
Deamidation (N-term) + Loss (17.02Da)
Water Loss (D, E, S, T) + Loss (2 of these, at 18.01Da each)
Dehydrated (C-term) + Loss (18.01Da)
One setting that you should probably change is "Max neutral losses". You have that set to 5 on:
Settings > Peptide Settings > Modifications
The problem with having it set that high is that it takes a really long time for Skyline to display to you the possible transitions for a precursor.
That is, in the Targets Tree, if you select the precursor ELASGLSFPVGFK/680.736++(heavy), and choose "Pick Children", it takes an enormous amount of time for the window to come up. Skyline is going through all of the possible combinations of 5 neutral losses. |
|
| |
| jfoe responded: |
2018-09-18 12:10 |
Thanks for the fast response!
The problem doesn't seem to have anything to do with always including losses vs only with library.
Restricting the use of too many losses would probably work in this case but doesn't get to the core of what I would suggest is behavior that is undesirable.
I set up an empty document with only matching library losses.
I will include the assay library that I am trying to import in a zip.
Maybe you could quickly try that.
What you will see is the result from the screenshot in the original post.
The peptide ELASGLSFPVGFK has 6 transitions, but they are not the same 6 transitions as given in the specral library, even though they were both defined by the assay library import in the same step. |
|
| |
| Nick Shulman responded: |
2018-09-18 12:56 |
Oops. It looks like Skyline has a bug in it.
When importing a transition list, Skyline tries to figure out what ion type each of product m/z's corresponds to. We have code that is supposed to say that any transition that has no losses is a better match than a transition that has losses. Unfortunately, we got the logic backwards, and so Skyline always prefers transitions that have losses.
I'll fix this. |
| |
| jfoe responded: |
2018-09-18 13:34 |
I also have the spectral library tolerance set to 0.001 so I am not sure why it even picked that one.
Does this tolerance apply to assay library imports?
I think it would be really bad to get wrong transitions after an import so it would be good to be able to have a strict tolerance. |
| |
| Nick Shulman responded: |
2018-09-18 13:56 |
When importing a transition list, the number that matters is the "Method match tolerance m/z". That setting is on:
Settings > Transition Settings > Instrument
You still have that setting set to its default value 0.055.
Your "Ion Match Tolerance" on "Transition Settings > Library" is .001, but that only affects things like choosing ions from a spectral library. |
| |
| Brendan MacLean responded: |
2018-09-18 15:33 |
The "Method match tolerance m/z" is supposed to control numbers likely to have been created by a person and entered into an instrument method. This was certainly the case for most transition lists from SRM imported into Skyline. The "Ion Match Tolerance" is supposed to be to control matching m/z values measured by a mass spectrometer, i.e. a spectral library. I do see the point that Assay Libraries blur these lines quite a bit, and though they are like Transition Lists, they also get called Spectral Libraries themselves and the source of the m/z values is nearly always instrument measured mass spectra.
Since, we now have different menu options for File > Import > Transition List and Assay Library, I suppose it might be appropriate to use "Ion Match Tolerance" during the Assay Library import. Hmmm... Something to think about. It might also add to confusion and lead to needing to ask when using File > Import > Transition List.
This is actually a very tough problem that has crept up on us as the popularity of these tabular Assay Library files has grown.
Thanks for bearing with us while we work through your issues and consider how we might make this easier, and thanks for helping us find that bug.
--Brendan |
| |
| jfoe responded: |
2018-09-19 04:55 |
OK then thank you for the help! |
| |
|
|
 Clipboard01.png
Clipboard01.png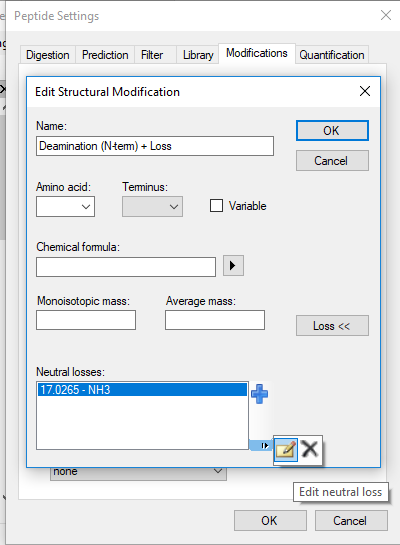 EditNeutralLoss.png
EditNeutralLoss.png