| Peak areas | Michael Cundell | 2018-09-10 14:24 | |||||||||||||||
Hi all, I am using an Orbitrap Fusion Lumos to acquire both DDA and PRM data within a single method. I became confused as to why Skyline v4.1.0.18169 was giving me spikey XIC's (see skyline.emf). These spikes are not present when I extract the same peptides transitions using Thermo's Freestyle application (Freestyle.jpg). Within freestyle I typically do this by selecting the appropriate PRM filter e.g. "FTMS + c NSI d Full ms2 123.4567@hcd28.00 [120.0000-1500.0000]" and then filling in the transition masses that I am interested in. This essentially ignores the DDA data within my RAW file. I think the spikes in the Skyline data are coming from the DDA part of the data. The DDA part within the method uses different parameters to the PRM part (eg 240ms fill time vs 120ms fill time respectively). In freestyle if I extract one of the transitions with the filter set to "ms2" (this includes both DDA and PRM data) you can see the source of the spikes in skyline comes from the DDA data (see Freestyle_2.jpg). Is there a way I can get skyline to ignore the DDA data within my RAW file? Thanks for your help. Best, Michael |
|||||||||||||||||
| |||||||||||||||||
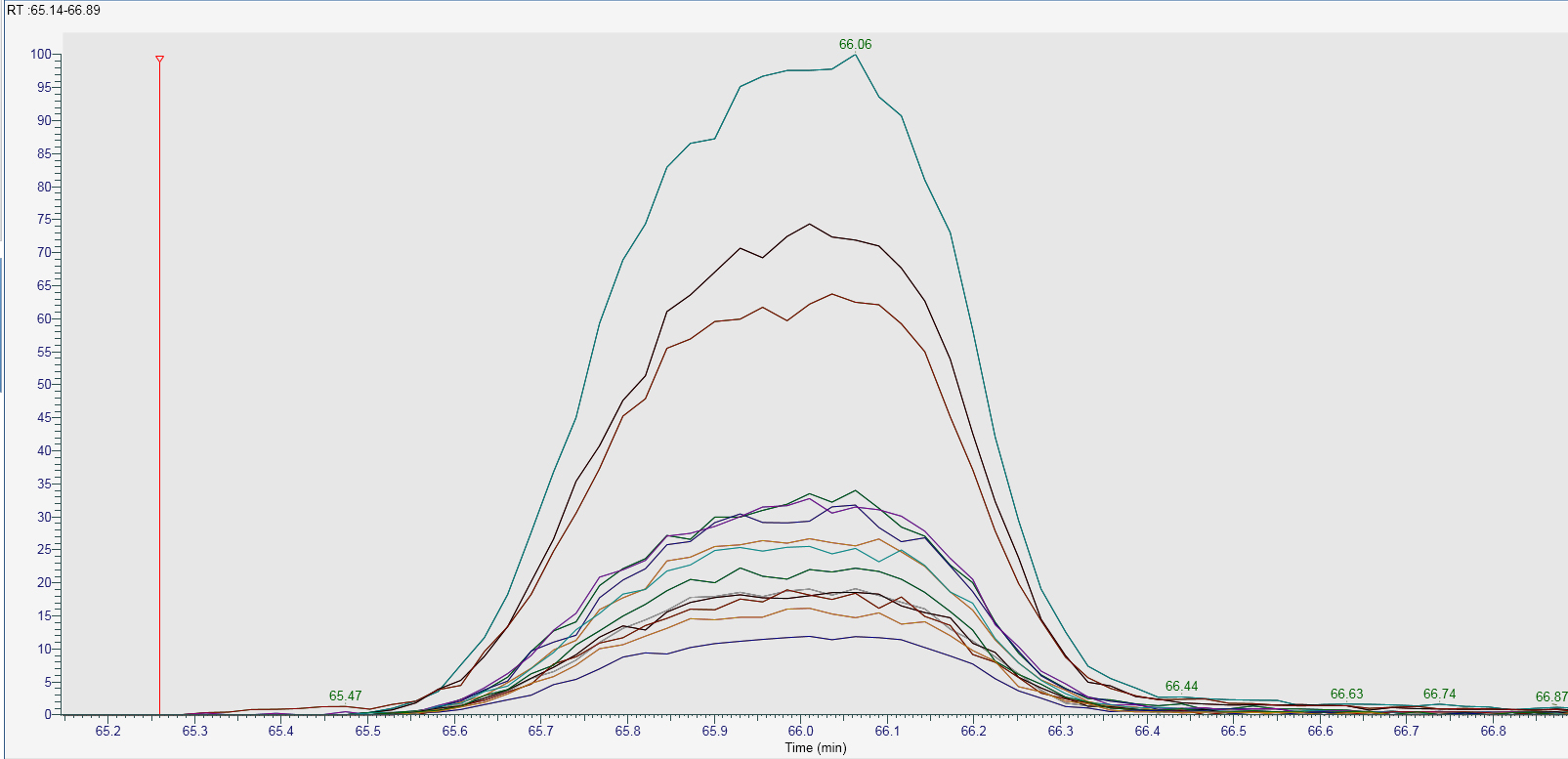 Freestyle.jpg
Freestyle.jpg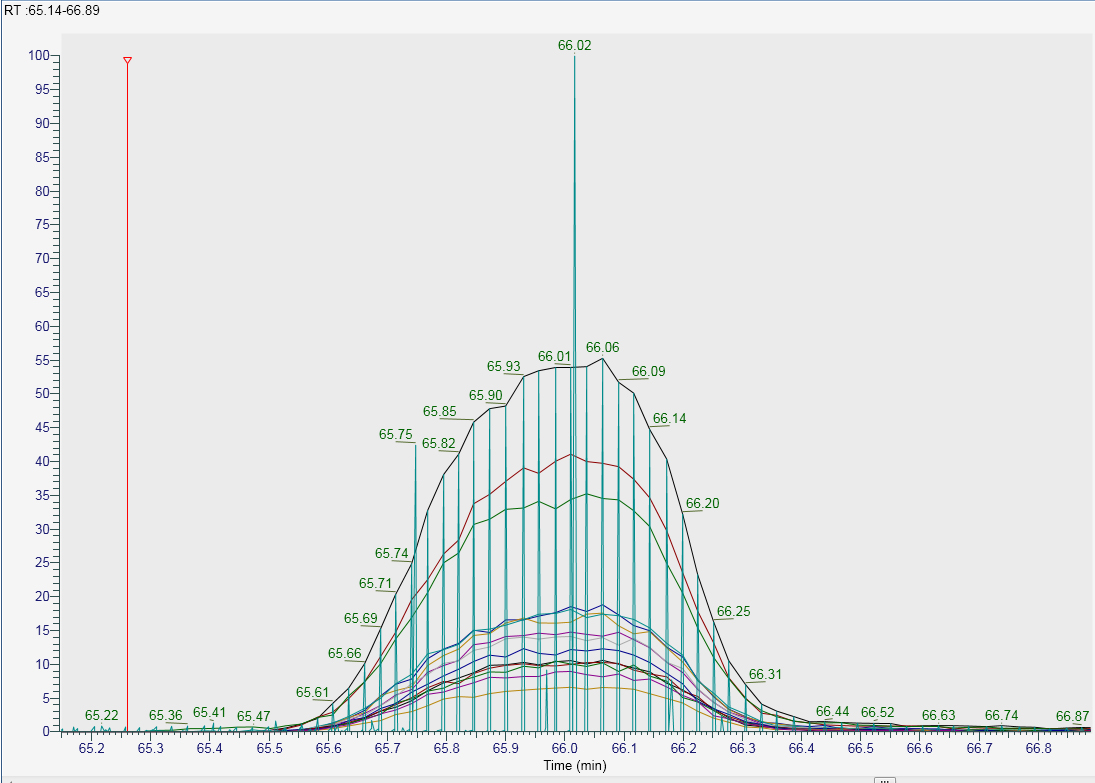 Freestyle_2.jpg
Freestyle_2.jpg