| |
| Brendan MacLean responded: |
2018-09-04 13:04 |
Hi Jakub,
They do have a meaning if you use a mechanism that imports protein sequences into the document, e.g. File > Import > FASTA, Edit > Insert > FASTA, Edit Insert > Proteins, or any of the direct Edit > Paste equivalents. If, however, you use View > Spectral Libraries - Add or Add All (with our without Associate Proteins) then you mostly bypass the enzyme cleavage except in how Skyline estimates the number of missed cleavages and what Skyline tells you about the added peptides not matching your filter settings. But you can add just about anything your mass spectrometer can measure (and for which Skyline understands the modifications) through View > Spectral Libraries - Add or Add All.
Interesting enzyme combination LysC and Tripsin. Amazingly, yours is the first request to mention it. What we have now solved other multi-enzymatic cleavage requests, primarily the ability to have both C- and N-terminal cleavage in the same definition. But, it is a very good point that mixing proteases can cause what you describe where you get cleavage always after some amino acids and that our "Unless followed by" option may only apply to a subset of the cleavage residues. Though, this is not the first example of a protease regimen that our current Enzyme setting doesn't support.
It may be that the best we can offer is:
- Use "Trypsin/P" as the Enzyme and live with overreporting of missed cleavages on R.
- Use "Trypsin" with View > Spectral Libraries - Add or Add All and live with underreporting of missed cleavages on K followed by P.
To me, #2 seems like it would have the last impact on how you interpret the results, but it also means you are limited to a certain way of adding your peptides, and you will need to build a background proteome if you want protein association with your peptides. For #1 to work, you may need to increase what you allow in Skyline for missed cleavages, but presumably in the end you are just looking to match peptides with the spectra that Mascot matched, and that should still work, just with some missed cleavages reported where they can't actually have occurred.
Hope this helps. Thanks for the feedback and explanation of what you would ideally like to see from Skyline.
--Brendan
|
| |
| Jakub Cervenka responded: |
2018-09-05 14:38 |
Hi Brendan,
Thank you very much for your reply! I will try both ways you suggested and will see, which one works better. I am quite surprised that nobody asked you for the LysC and Trypsin combination before, as it is not so rare combination and even some manufacturers sell these enzymes as a mix. It would be nice to have a possibility to use this combination directly in Skyline or have an option to set the parameters correctly – maybe in one of future version? :-)
Thank you again for your help and for the Skyline, of course. I really appreciate that the Skyline (especially daily version) is up-to-date and user-friendly software with perfect professional support from you and other people from Skyline team. Thanks!
Jakub
|
| |
| Jakub Cervenka responded: |
2018-09-06 07:08 |
Hi Brendan,
We tried both possibilities you mentioned, but the second option – “Use "Trypsin" with View > Spectral Libraries - Add or Add All and live with underreporting of missed cleavages on K followed by P” is not working. Even though we set proteome background (the same FASTA as for Mascot search) and maximum missed cleavage (2), no new proteins appeared after adding all peptides from spectral library. Moreover, all new peptides (approx. two thousands) were associated to “Library peptides” instead of proteins. After deletion of the Library peptides icon all newly added peptides were gone.
What could be the reason for the problem with association of new peptides to proteins? May be the problem in FASTA file (contains contaminants and randomized decoys)?
The other option (the first one) you suggested worked and we got several hundreds of new peptides, but it is only approx. 1/3 of peptides added by the second way.
Thank you,
Jakub
|
| |
| Brendan MacLean responded: |
2018-09-06 07:45 |
I think you will need to provide us your files. You can use File > Share - Complete to save the Skyline document (with spectral library and background proteome) to a .sky.zip file and post that to:
http://skyline.ms/files.url
Unless for some reason you are using an older version of Skyline or older background proteome. The ability of a background proteome to recognize arbitrarily cleaved peptides as matching any protein that contains their sequence is relatively new.
If you are using Skyline 4.1 or Skyline-daily, then we will need to take a closer look at why it is failing to behave as expected.
--Brendan
|
| |
| Jakub Cervenka responded: |
2018-09-10 06:54 |
Hi Brendan,
I am sorry, I found that it was our mistake, because we did not checked the option “Associate proteins”. When I did it, peptides were added to proteins and it worked better than our old way of FASTA import. Thank you!
One last question – I see different numbers of peptides in my library in Skyline (in Spectral Library Explorer) than in finished Skyline (even when filtering is taken to count – see attached files). Why is this happening?
Thank you for your time and help!
Jakub
|
|
| |
| Brendan MacLean responded: |
2018-09-10 08:11 |
Hi Jakub,
Sorry for the confusion. The number reported in the Spectral Library Explorer as "peptides" (9672) should actually be "precursors" because it counts multiple charge states of the same peptide sequence (and also multiple isotope labeled states), whereas these will be counted as a single (modified) peptides sequence in Skyline's peptide counts (in the status bar and in the final message box you show). So, you should be comparing precursors with precursor, which means you start with 9672 and end with 8572. So, there are 1100 fewer precursors than found in your library.
I suspect that Skyline is really talking about peptides when it tells you that 1020 match multiple proteins and it may also be talking about peptides when it says that 18 do not match your filter settings, but I will have to look more closely at that.
So, I think we need to do a better job of rationalizing these numbers to make it easier on you doing straight math and knowing which numbers to compare. Because, clearly you can't do straight math with any of the numbers to understand what Skyline has done, i.e. 1020 + 18 + 1 = 1039 which is 61 precursors less than the reported final difference.
But, I am pretty sure this is related to a difference in the meaning of "peptides" in the various numbers getting reported. There are actually 9672 precursors in your library. If you click on the "..." (ellipsis) button next to the library name in the Library Explorer, you will see the number of "Unique peptides" reported as well as the number of "Unique precursors". The latter should match the number you see a "Peptides 1 through 100 of total 9672", while the former should be a smaller number, but if you have any isotope labeling, then even the "Unique peptides" reported here may report numbers that will differ from what you get when you add everything to the Targets view.
Anyway, it is complicated. I will look for ways to be clearer about reporting "precursors" which are the only numbers we can be completely confident of in this context.
Thanks for your post to the Skyline support board.
--Brendan
|
| |
|
|
 Library_import_filtering.PNG
Library_import_filtering.PNG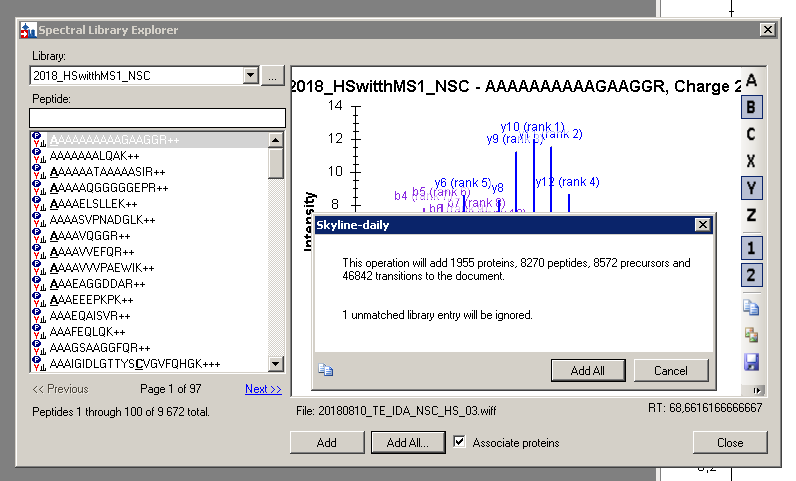 Library_import_finished.PNG
Library_import_finished.PNG