Which feature scores are unavailable that you were hoping to be able to use in your model?
In the screenshot that you provided, there are some scores (e.g. "Shape (weighted)") where the feature is not being used because you have unchecked the checkbox next to them. For those scores, you could check the checkbox and press the "Train Model" button and that feature will be used.
Some other feature scores are grayed out and in italics (e.g. "Retention time difference"). Those are features for which there is at least one peptide in your document that does not have a value for that feature. You can find the peptides which are missing that feature by doing the following:
1. In the "Edit Peak Scoring Model" dialog, click on one of the cells in the feature row in the "Available feature scores" grid.
2. Choose the "Feature Scores" tab.
3. If the feature is missing from some but not all of your peptides, then when you hover the mouse below the "unknown" bar on the bar graph, a binoculars button will appear and you can click it to get a list of all of the peptides that are missing that feature.
I do not understand your question about exporting protein lists and group comparison. Can you maybe send us your Skyline document?
In Skyline, you can use the menu item:
File > Share > (complete)
to create a .zip file containing your Skyline document and supporting files, including extracted chromatograms.
You can upload that .zip file here:
https://skyline.ms/files.url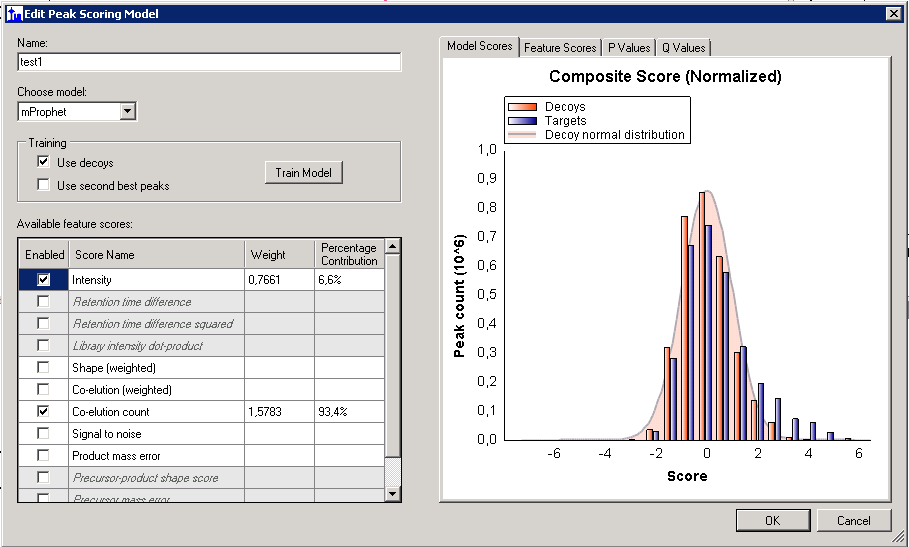 PeakPickingModel.PNG
PeakPickingModel.PNG