| Brendan MacLean responded: |
2018-05-09 10:06 |
Hi Keri,
I think we will need to see your document to get a better idea of what you are trying to achieve. You can use File > Share - Complete to save it to ZIP file and post it to:
http://skyline.ms/files.url
Or if the ZIP file is relatively small, you can just post it directly to this thread.
We'll do our best to help you achieve your desired experimental setup.
--Brendan |
| |
| Keri responded: |
2018-05-09 10:32 |
Thanks Brendan,
The files were too large to upload on the thread. I've attempted to upload the zip'd skyline directory to the link that you sent, but it gives me the following error:
"You do not have privileges to this directory. Verify that you are signed in appropriately."
I'm signed in w/ username and password. Is there somewhere else that I need to log-in to be able to upload the files?
Keri |
| |
| Keri responded: |
2018-05-09 10:35 |
It looks like, despite the error, the file may have uploaded. It's called skyline_test.zip
Thanks for the help!
Keri |
| |
| Nick Shulman responded: |
2018-05-09 13:05 |
Thank you for sending us your files. Our website unfortunately gives you the "no privileges" error if you try to upload the same file more than once, rather than telling you that the file already exists. We hope to fix that error message soon.
Currently you have three different "structural" modifications defined: C+57, C+521, and C+527.
Are you saying that all Cysteines that have the C+521 modification on them also have the C+57 modification on them? If that is the case, then I would recommend that you define a single modification which is C+578. It is theoretically possible in Skyline to have two different structural modifications on the same amino acid, but it is really tricky to get Skyline to add such a peptide to your document, so we recommend that you combine the two modifications into a single modification.
Your C+521 and C+527 modifications really are just isotopic variants of each other, right? In that case, you should define an Isotope Modification for that 6 Dalton difference. It looks like the chemical formula of that modification would be:
C'5N'-C5N
That is, you add 5 heavy carbons and one heavy nitrogen and subtract 5 regular carbons and 1 regular nitrogen.
I think you should make both of your structural modifications into "explicit" modifications. That means that you uncheck the checkboxes next to them in:
Settings > Peptide Settings > Modifications
and then you explicitly add them to the peptides that should have them by using the "Edit > Modify Peptide" menu item.
I have attached a .sky.zip file where I gave modifications to your first two peptides in the way that I think they should be.
You might also find some helpful information in Webinar 10 "Working with modifications in Skyline":
http://skyline.ms/webinar10.url |
|
| |
| Keri responded: |
2018-05-09 17:18 |
Hi Nick,
Thanks for the information. That is quite helpful. I previously tried to have the two modifications separate (+57 and +464), but as you mention, that did seem quite challenging to execute, so all cysteines in the dataset are either +57, +521 or +527. There should never be +584 or other additive masses on individual cysteines. And, yes the modifications are just isotopic variations of each other, so a +6 modification would be fine. I'm not sure whether the edit>modify peptide will work, as I really want to be able to process the whole dataset rather than individual peptides. Is there a way to batch edit peptides? Also most peptides will show up as peptide pairs (+521, +527), so I would like to be able to report the H/L ratios of pairs of peptides across all peptides that contain those modifications. Hopefully these is a way to do this in skyline?
Thanks again
Keri |
| |
| Nick Shulman responded: |
2018-05-09 17:52 |
One thing that you can do is:
Edit > Insert > Peptides
and you can specify a modified sequence in the "Sequence" column such as:
EGLLLWC[+521]QR
However, it sounds like you also want to tell Skyline that every C[+521] should also get a matching C[+527]. There is not really a way to say that the +6 modification applies to C[+521] but does not apply to C[+57].
Instead, what I think you should do is create two separate Skyline documents like I have attached "labeled.sky" and "unlabeled.sky".
In "labeled.sky" I told Skyline that every C should have a static modification "+521", and there should also be a heavy variant "+6".
In "unlabeled.sky" I told Skyline that every C should have the static modification "+57" and there is no heavy variant.
Then, you can open up "unlabeled.sky" and use the menu item:
File > Import > Document
and import "labeled.sky" into it, and you will get a merged document with two different sorts of peptides in it.
If you do "Edit > Insert > Peptides" into either of these documents and paste in unmodified sequences, you will end up with peptides with the right modifications on them. And then you can merge the documents to create a single document with all of the modified peptides that you want.
Do you have any peptides with more than one Cysteine in them where one of the Cysteines is +57 and the other is +521? If so, then you will probably have to manually tell Skyline about the modifications. |
|
| |
| Keri responded: |
2018-05-09 18:19 |
Thanks Nick,
There are definitely peptides in the dataset with more than one cysteine where one is +57 and one is +521. There are also cases where peptides contain +527 but there is no equivalent +521 peptide. I'd really prefer to not have to manually process all of the peptides containing multiple cysteines,, as we have quite a few datasets in this format. But if there is no other way, I will try that as a starting point.
For your suggestion with the labeled and unlabeled documents, I was unable to combine your files like you suggested, as when I import the labeled.sky file it gives the following error: System.IO.InvalidDataException: The modification 'TEV' already exists with a different definition. ---> System.IO.InvalidDataException: The modification 'TEV' already exists with a different definition. This happens even when I start with a blank skyline document. Hopefully something easy to fix.
Thanks
Keri |
| |
| Nick Shulman responded: |
2018-05-09 21:15 |
I think that "ACouplePeptides.sky.zip" that I gave you earlier gave you a different definition for the modification "TEV" which is messing you up now.
Here's what you need to do in Skyline-Daily:
1. File > New
2. Tools > Options > Miscellaneous > Clear All Saved Settings
3. Open "unlabeled.sky"
4. File > Import > Document and choose "labeled.sky" |
| |
| Keri responded: |
2018-05-10 11:39 |
Got it. They open now. Thanks! This seems like it could be a good solution for all the peptides that only contain one cysteine, but I'd still love to figure out a more permanent solution for those peptides that contain multiple cysteines. How does Skyline handle other PTMs where there are multiple possible modifications, for example H/L mono-, di- and trimethyl lysine? |
| |
| Nick Shulman responded: |
2020-10-23 13:37 |
Keri,
I cannot think of a way to give you the control that you need over which cysteines get the heavy modification applied to them. Maybe someone else on this support board will have a good idea.
To summarize, all of your light cysteines have either the usual [+57] modification applied to them or the [+521] modification.
Your heavy peptides need are formed by changing all of the [+521] to [+527] and leaving the [+57] cysteines alone.
I cannot think of any way to accomplish this. There might be some way to do this by doing "Edit > Insert > Peptides" or "Edit > Insert > Transition List", but I cannot figure one out.
(Also, the new Skyline-Daily feature "Permute Isotope Modifications" seemed like it might work, but introduces a bunch of other problems).
Hopefully someone else has a good idea, or it might be that we should implement new features to support this, such as allowing you to specify both the heavy and light modified peptide sequences when importing a transition list.
-- Nick |
| |
| Brendan MacLean (test) responded: |
2020-10-23 16:07 |
You can achieve this with Edit > Insert > Peptides. I did with the following added to my document:
1. Normal "Carbamidomethyl (C)" = [+57]
2. A structural modification I called "Tagged (C)" = [+521]
3. A isotope labeling modification which I called "Labeled Tagged (C)" = +6
This allowed me to add the following text through Edit > Insert > Peptides:
PEC[+57]PIC[+527]ER
Which gave me targets like this:
PECPICER
762.7204++
precursor - 762.7204++
P [y5] - 1138.3076+
I [y4] - 1041.2548+
C [y3] - 928.1707+
765.7204++ (heavy)
precursor - 765.7204++
P [y5] - 1144.3076+
I [y4] - 1047.2548+
C [y3] - 934.1707+
Is this what you had in mind? I am pretty sure what you want is achievable even if this is not it. The ModificationMatcher responsible for handling modifications in Edit > Insert > Peptides is extremely flexible.
--Brendan |
| |
| Keri responded: |
2020-10-23 16:20 |
Thanks Brendan---In principle that is what we are looking for, though I would love to avoid having to add the peptides manually. Each experiment will have 15-20k of these types of peptides, so am trying to automate as much of this workflow as possible.
Thanks for looking into this.
---Keri |
| |
| Brendan MacLean responded: |
2020-10-23 19:52 |
Skyline should use the same ModificationMatcher when adding peptides from a DDA search engine. So, as long as the search engine provides matches with modified peptides that look like what I provided, or actually, you should be able to match both light and heavy peptides. So, if the search results contained matches like either:
PEC[+57]PIC[+527]ER
or
PEC[+57]PIC[+521]ER
Skyline would automatically match these correctly to the modifications I have suggested you add to your document.
If you need more help with this, it would help us a lot to see one of these 15-20K spectral libraries with matches like this. |
| |
| Keri responded: |
2020-10-25 22:26 |
Thanks Brandon,
Attached is the list of peptides for this experiment, as well as the mzid output from MSGF+ and the mzXML file. When I try to paste this list into the insert peptide window per these instructions, Skyline freezes. If I just paste a few entries, there's no issues, so I think the freezing may be due to the size of the list.
We typically search with +57 as a structural modification and then +464 or +470 as variable mods, as that gives us more IDs compared to search with +57 as an additional variable mod. This means that our output looks like PEC[+57]PIC[+57+464]ER. For the text file below, I cleaned up the data to make it look like your example.
Keri |
| |
| Nick Shulman responded: |
2020-10-25 22:48 |
Keri,
You did not actually attach a file to your last message. Can you try again?
If the file you are attaching is more than 50MB (which would be an implausibly large list of peptides) you would need to upload the file here:
https://skyline.ms/files.url
-- Nick |
| |
| Brendan MacLean responded: |
2020-10-26 08:59 |
Interesting. Yeah, getting the mzid and mzXML files would be great. Are you able to build a spectral library from them? I would not expect any of our software to recognize the C[+57+464] modification format. If that is natively what MSGF+ produces, then probably we should do our best to support it. It would also be good to get your list and fix Skyline to show a progress bar, and ideally improve performance, for your adding that very large peptide list.
If we can get your files, I am sure we can help you come up with a usable workflow that will allow you to achieve what you are aiming for.
Thanks for providing details and helping us work through this issue.
--Brendan |
| |
| Nick Shulman responded: |
2020-10-26 09:14 |
Keri,
When you are pasting a list of modified peptide sequences into Skyline, one thing which can cause it to take a really long time is if you have a lot of structural and/or isotope modifications in the lists at:
Settings > Peptide Settings > Modifications
When Skyline sees a modified sequence with modification masses in it (e.g. "C[+527]"), Skyline has to figure out what combination of structural and isotope modifications might have been applied to that residue to result in that mass. The amount of time that this can take may increase exponentially with the number of modifications in the lists on the Modifications tab of the Peptide Settings dialog.
If you are finding that pasting a list of modified sequences is hanging, you should remove all unnecessary modifications from the Peptide Settings Modifications lists, so that Skyline does not need to consider as many modifications.
-- Nick |
| |
| Keri responded: |
2020-10-26 11:01 |
I did finally get the paste to work. mzid and mzXML have been uploaded. And yes, I can generate spectral libraries with the C[+57+464] modification format seemingly without issues, which is surprising.
Keri |
| |
| Keri responded: |
2020-11-12 21:14 |
Hi Nick,
Just wanted to circle back and see if you'd gotten those files I uploaded and whether the modifications worked in your hands? I am still struggling to resolve this problem.
Thank you!
Keri |
| |
| Keri responded: |
2020-11-12 21:33 |
One more point of clarification--I have modified the search to include three variable mods (+57, +521 and +527). Skyline handles this data for peptides that contain single cysteines well, but for those that contain multiple cysteines, the +6 heavy label is added onto both the +521 and +57 to give an output like the following PEPC[+63]PEPC[+527]. As it's not possible to add +6 onto the +57, these multi-cys peptides (about 10% of the data) would fail quant.
Keri |
| |
| Nick Shulman responded: |
2020-11-14 09:35 |
Keri,
Thank you for sending us your .mzid and .mzml files on Oct 26.
I am not sure what I am supposed to be looking at.
I see that as you said your mzid file searched for cysteine modifications +57, +464 and +470, where the +57 is applied to every Cysteine and the +464 or +470 gets applied to some Cysteines such that they become either +521 or +527.
This does not matter to Skyline. By the time the search results get put into the .blib file, they have lost all information about the individual modifications, so Skyline only sees the total mass difference (+57, +521 or +527).
It looks like your peptide search results that my understanding cannot physically exist. For instance, there's the peptide C[+521]KTC[+527]SEPK+++. That must be a misidentification since you would never have +521 an +527 in the same peptide.
I don't think there is going to be any way to tell Skyline the rule that C[+521] should be changed to C[+527] for the heavy, but C[+57] should be left alone. It was possible to make this work for single Cysteine containing peptides by doing the "File > Import Document" above, but for peptides with two different types of Cysteines in it, you really will need to right-click on each peptide in the Targets tree and choose "Modify Peptide".
I could imagine a way to do this where you repurpose some other amino acid (such as Isoleucine) and give that modifications so that it has the same chemical formula as C[+527]. But that might cause a different set of problems.
I might be able to write a script which takes a list of peptides and produces a .sky file with the right set of modified peptides. I will investigate whether that is feasible.
-- Nick |
| |
| Keri responded: |
2020-11-14 10:17 |
Thanks Nik---ignore the mix of 521/527 peps for now. They are obviously false calls and can be filtered out. As you highlighted the issue is telling Skyline that C[+521] should be changed to C[+527] for the heavy, but C[+57] should be left alone. If there is a way to make this happen, that would solve most of our quant problems.
Keri |
| |
| Nick Shulman responded: |
2020-11-15 11:04 |
Keri,
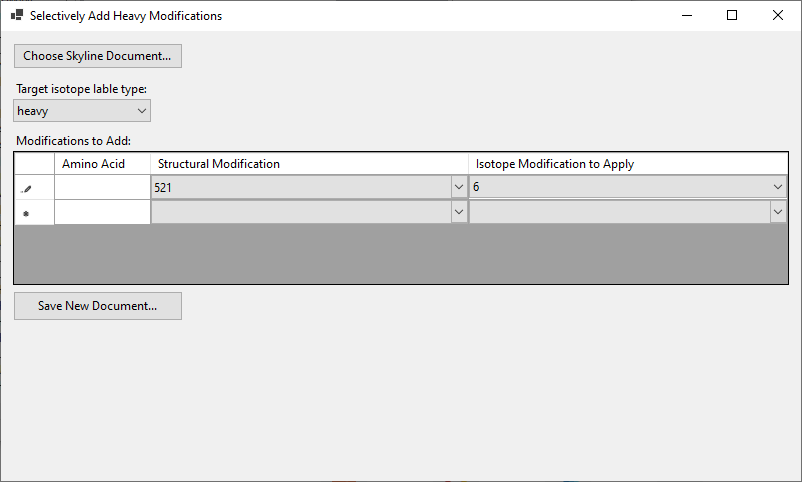
I have written a tool which will apply a heavy modification to only those residues which already have a particular structural modification on them.
You can install the tool from here:
https://proteome.gs.washington.edu/~nicksh/kbackus/AddLabelType/setup.exe
The tool lets you open up a .sky file, and you can specify that a particular heavy modification should only be added to a residues that already have a particular structural modification on them. (see attached screenshot).
Then, you can use the "Save New Document" button to write out a new .sky file (I recommend overwriting the .sky file that you originally opened, so that it will be right next to all the other files it needs such as the .skyd file).
When you open this new document up in Skyline, all of the heavy precursors will have been removed, but Skyline will know which modifications those heavy precursors are supposed to have. You can then use the menu item in Skyline:
Refine > Advanced
and click the "Add" checkbox next to "Remove Label Type" and tell Skyline to add the Heavy label type.
I have not tested this tool very much, but it will probably work for you.
I have also uploaded a .sky.zip which is what I got when I used the tool on your file "JCC1_TPP.sky.zip". You can download what I have created here:
https://proteome.gs.washington.edu/~nicksh/kbackus/JC1_TPP_fixed.sky.zip
-- Nick |
|
| |
| Keri responded: |
2020-11-15 19:02 |
|
| |
 AddLabelTypeTool.png
AddLabelTypeTool.png