| Problems (PRM workflow and isotope tags targeting cysteine residue) | ylam | 2018-04-24 14:20 | |||||||||||||||||||||||||||||||||||
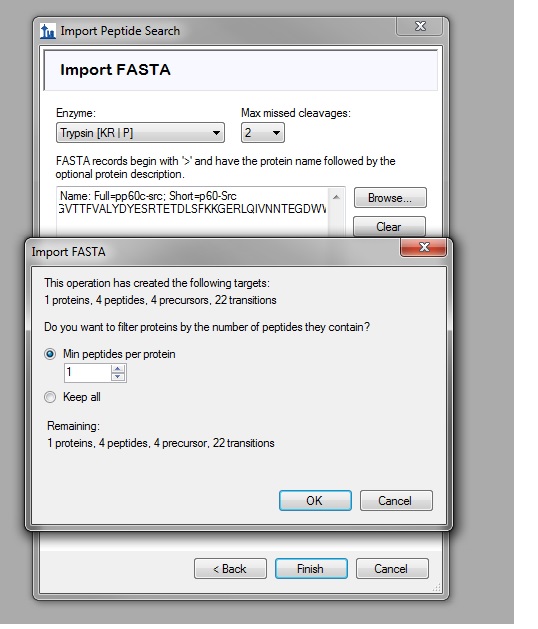
Hello: I have a question regarding Skyline and am wondering if you could help me out. I used Skyline a couple of years ago, and it interpreted the data very well for one of our studies. Thanks for your help at that time! 1) I am now trying to use Skyline again for another study, and I am having several problems/questions. I am using the PRM workflow and trying to import the Proteome Discoverer 1.4 msf file into the Skyline. When I input the sequence of the protein of interest with trypsin as an option (Import FASTA), it says the protein only has 4 peptides (see attached: SkylineQuestion.jpg). However, the protein clearly has more than 8 peptides, how come? The sequence is listed below: >sp|P12931.3|SRC_HUMAN RecName: Full=Proto-oncogene tyrosine-protein kinase Src; AltName: Full=Proto-oncogene c-Src; AltName: Full=pp60c-src; Short=p60-Src MGSNKSKPKDASQRRRSLEPAENVHGAGGGAFPASQTPSKPASADGHRGPSAAFAPAAAEPKLFGGFNSS DTVTSPQRAGPLAGGVTTFVALYDYESRTETDLSFKKGERLQIVNNTEGDWWLAHSLSTGQTGYIPSNYV APSDSIQAEEWYFGKITRRESERLLLNAENPRGTFLVRESETTKGAYCLSVSDFDNAKGLNVKHYKIRKL DSGGFYITSRTQFNSLQQLVAYYSKHADGLCHRLTTVCPTSKPQTQGLAKDAWEIPRESLRLEVKLGQGC FGEVWMGTWNGTTRVAIKTLKPGTMSPEAFLQEAQVMKKLRHEKLVQLYAVVSEEPIYIVTEYMSKGSLL DFLKGETGKYLRLPQLVDMAAQIASGMAYVERMNYVHRDLRAANILVGENLVCKVADFGLARLIEDNEYT ARQGAKFPIKWTAPEAALYGRFTIKSDVWSFGILLTELTTKGRVPYPGMVNREVLDQVERGYRMPCPPEC PESLHDLMCQCWRKEPEERPTFEYLQAFLEDYFTSTEPQYQPGENL 2) It looks like the Skyline was not able to incorporate all the peptide identification from Proteome Discoverer results, although I have set the cut-off score = 0, when I imported the results. 3) We are using a cysteine-reactive tag C(8)H(10)O(2) [138.06808 Da] and its isotope (deuterium) counterpart 2H(6)C(8)H(4)O(2) [144.105740 Da] for our experiments. I have set these two as variable modification and 2H(6)C(8)H(4)O(2) - C(8)H(10)O(2) = 6.03766 Da as "heavy isotope modification". But Skyline seems to also see 144.105740 Da + 6.03766 Da as "heavy" as well. It has been a while since I used the Skyline, any help will be greatly appreciated! Best wishes, Wai Ying Wai Lam VGN Proteomics Facility University of Vermont 802-656-4709 |
|||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
 SkylineQuestion.jpg
SkylineQuestion.jpg