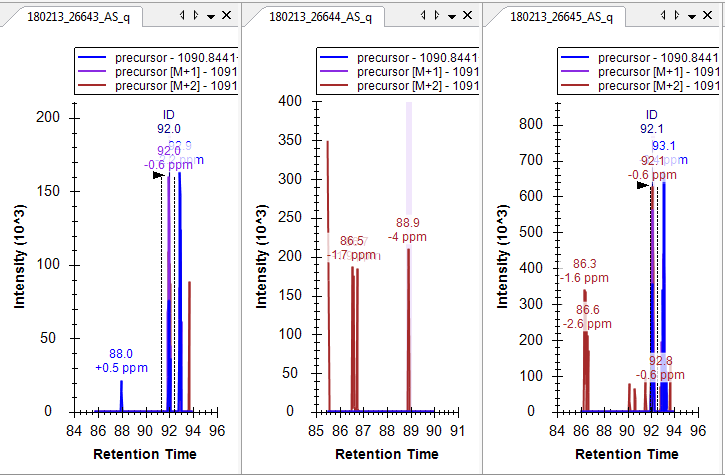
Hello Skyline team, we are performing MS1 quant using the new version of Skyline (4.1.11796) and have noticed that for some peptides the alignment goes wrong, leaving the peak outside of the window for one or more runs. This happens even when the peaks are close to the same RT when we look at XICs in Xcalibur. This was not happening in previous versions we have used (3.7 and prior). We even see this if we use much wider RT windows in 4.1, say 8 min instead of our typical 1-2min. I have attached one example, screenshots of Skyline and XCalibur for 1 peptide. Thanks! |
| |
| Nick Shulman responded: |
2018-03-16 11:40 |
Can you send us your Skyline document and a couple of your .raw files?
In Skyline, you can use the menu item:
File > Share > (complete)
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
You can upload that .zip file, as well as your .raw files here:
https://skyline.ms/files.url
The length of the chromatogram that Skyline extracts depends on the Retention Time Filtering settings on:
Settings > Transition Settings > Full Scan
Since you're talking about alignment, it sounds like you are using the option "X minutes from MS/MS IDs". |
| |
| Brendan MacLean responded: |
2018-03-17 17:45 |
It may also be useful to right-click in the chromatogram graph and choose Peptide ID Times > Aligned. This should cause Skyline to show where it is expecting the ID'd spectra from other runs to appear in the run without any.
However, I guess we should confirm that your are using the "X minutes from MS/MS IDs" option as Nick suggests, which doesn't seem possible to me, because it should cause the extracted chromatograms in the runs with IDs to be centered around those IDs. The IDs instead appear in the last 25% of the extracted chromatogram, indicating that some other method is being used to determine the location of the extraction window.
Yes, having the files to look at might help. I expect this is due to a difference in use, and not a difference between 4.1 and 3.7. Unless you have done a very thorough side-by-side test, it is much more likely that something changed about the way you processed your data with 3.7 versus now.
--Brendan |
| |
| mstokes responded: |
2018-03-18 18:21 |
Thanks guys, yes, using "X min from MS/MS ID." Typically set at 2min but we see issue even with 8-10min in new version. It is entirely possible that it is due to difference in use, though we checked peptide settings and transition settings side by side and didn't notice anything different. We will generate a file in 4.1 using some data that we can share and upload to you. Thanks again! |
| |
| mstokes responded: |
2018-03-20 07:35 |
Hi guys, uploaded a test file with 4 runs along with .raw files:
Skyline4_test1.sky.zip
180223_26525_AS_L.raw
180223_26526_AS_L.raw
180223_26527_AS_L.raw
180223_26528_AS_L.raw
I also found a few peptides with the issue, though these are just from the first few alphabetically, there will be more I'm sure:
AAATEDATPAALEKGTHNGNNPPTQPGLSPNGLNSGQMAN (missed 1)
AADTIGYPVMIR
AAGAQIQGMK
AALPSHVVTMLDNFPTNLHPMSQLSAAITALNSESNFAR
AALSAGKVPPETIDSVIVGNVMQSSSDAAYLAR (missed 1)
AANEAGYFNEEMAPIEVK
Thanks again for your help! |
| |
| Nick Shulman responded: |
2018-03-20 11:46 |
Can you post a screenshot of what you are seeing.
All of those peptides look fine to me, but I might not know what I am supposed to be looking for.
When you tell Skyline to only extract chromatograms within 2 minutes around your MS/MS ID's, the length of the chromatogram depends on where and whether the peptide was ID'd in the replicate.
If there is a peptide spectrum match for that peptide in that replicate, then the chromatogram will extend from 2 minutes before the first ID of that peptide in that file, to 2 minutes after the last ID.
If there is no peptide spectrum match for that peptide in that replicate, but the peptide was ID'd in other replicates, then Skyline does a retention time alignment of all of the IDs in all of the replicates. You can see this alignment by going to:
View > Retention Times > Alignment
You can right-click on the chromatogram and turn on both of the following:
Peptide ID Times > Matching
Peptide ID Times > Aligned
(see attached screenshot).
If there is a peptide ID in that particular replicate, it shows up on the chromatogram as dark blue, and the extracted chromatogram extends for 2 minutes either side of that ID.
If there was no peptide ID in that particular replicate, then the ID times from other runs show up as light blue lines, and the chromatogram extends from 2 minutes before the first light blue line to 2 minutes after the last light blue line.
What are you seeing that doesn't look right? |
|
| |
| mstokes responded: |
2018-03-20 12:02 |
Thanks Nick, I have posted a screenshot of skyline and xcalibur for one peptide. What I'm seeing in this case is that for the 2nd injection (26526) the RT range in skyline is out at ~73-77min even though the peptide ID in the other runs is ~66min. Skyline has aligned correctly for the 1st run (26525) and found the peak(s), but wasn't able to do so in 26526. Looking at XIC in xcalibur we see that the peak is present in all 4 runs and is very close RT-wise in all 4. Thanks! |
|
| |
| Nick Shulman responded: |
2018-03-20 12:54 |
Oh. I see what's going wrong.
When Skyline is trying to decide over what retention time range to extract chromatograms, Skyline is not paying attention to the peptide modifications.
That is, when Skyline looks for the retention times for:
AANEAGYFNEEM[+15.99491]APIEVK
Skyline is including the retention time for:
AANEAGYFNEEMAPIEVK
as well.
This is a bug, but, as far as I can tell, Skyline has behaved this way since when we first implemented the "retention time filtering" feature.
We did not notice this bug, because, usually, being more lenient about what Skyline considers a peptide match would only result in a longer chromatogram being extracted-- that is, the extraction range would contain both the modified and unmodified peptides.
However, for your replicate "180223_26526_AS_L", only the unmodified form of the peptide was identified.
Your other replicates did identify the modified peptide, however, because Skyline thought it found a perfect match for "AANEAGYFNEEMAPIEVK" in "180223_26526_AS_L" (which is at 74 minutes), Skyline does not do anything with the retention times of the modified peptide in your other replicate (which are at 66 minutes).
I hope this explanation helps. I will try to fix this bug in Skyline-Daily. |
| |
| mstokes responded: |
2018-03-20 12:58 |
Yes that is helpful and makes sense, thanks for finding that! If you are able to fix the bug in Daily will it eventually be fixed in a regular update of 4.1 as well? |
| |
| Nick Shulman responded: |
2018-03-20 13:55 |
I was mistaken that this has always been this way.
This bug was newly introduced in 4.1 as part of the feature where we store modifications in libraries with higher precision.
I am sure we will fix this in 4.1 and Skyline-Daily. |
| |
|
|
 Skyline_3run_1090.8441.png
Skyline_3run_1090.8441.png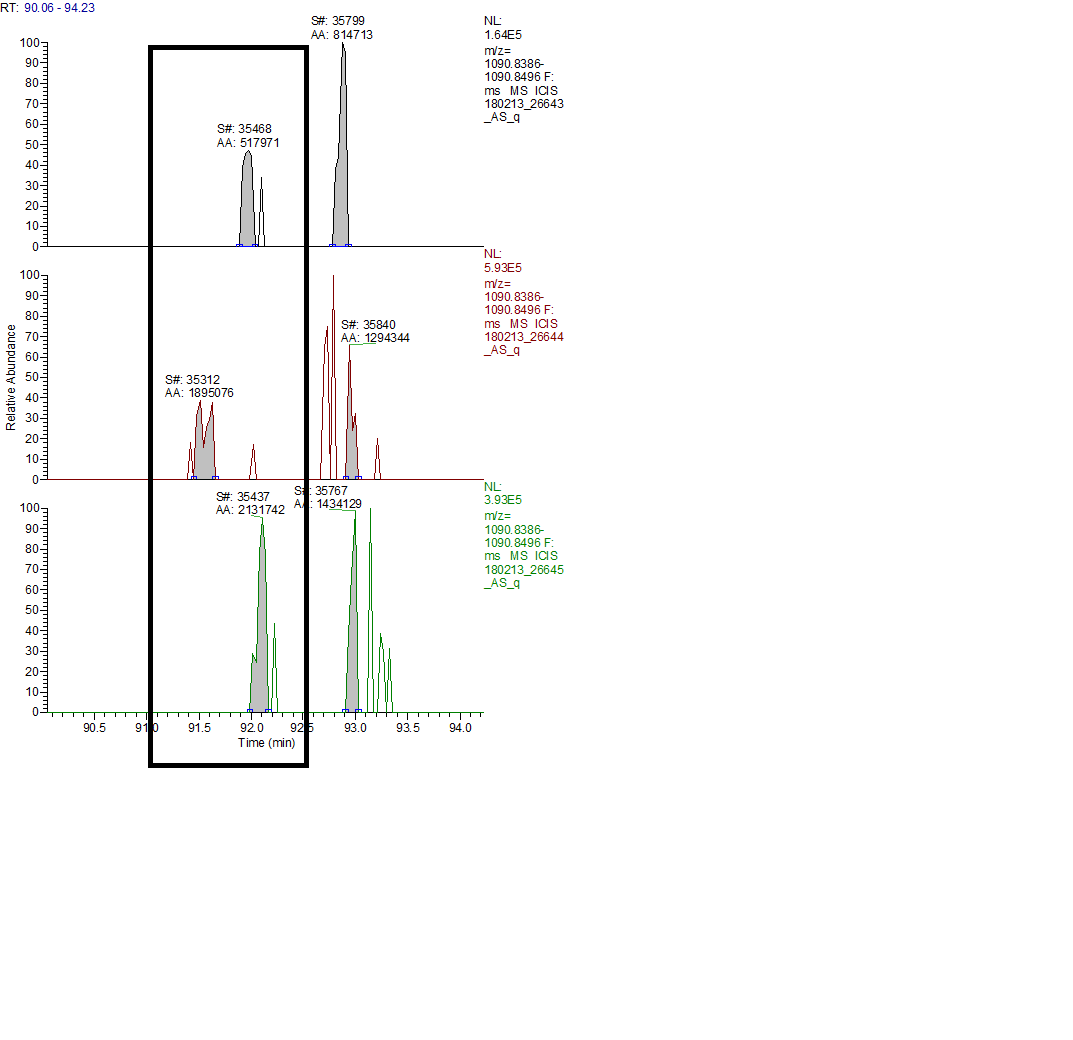 Xcalibur_1090.8441.png
Xcalibur_1090.8441.png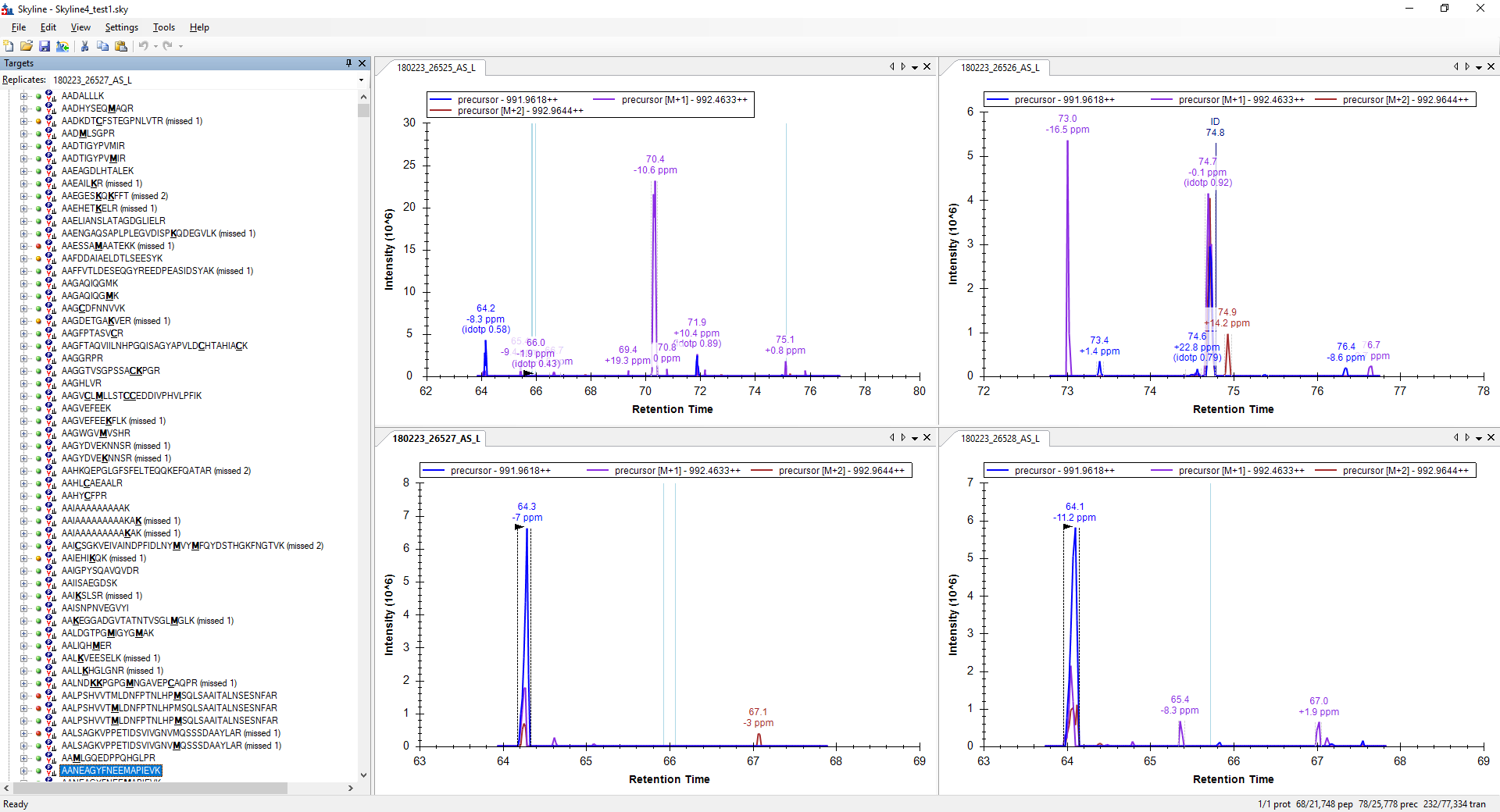 mstokes_screenshot.png
mstokes_screenshot.png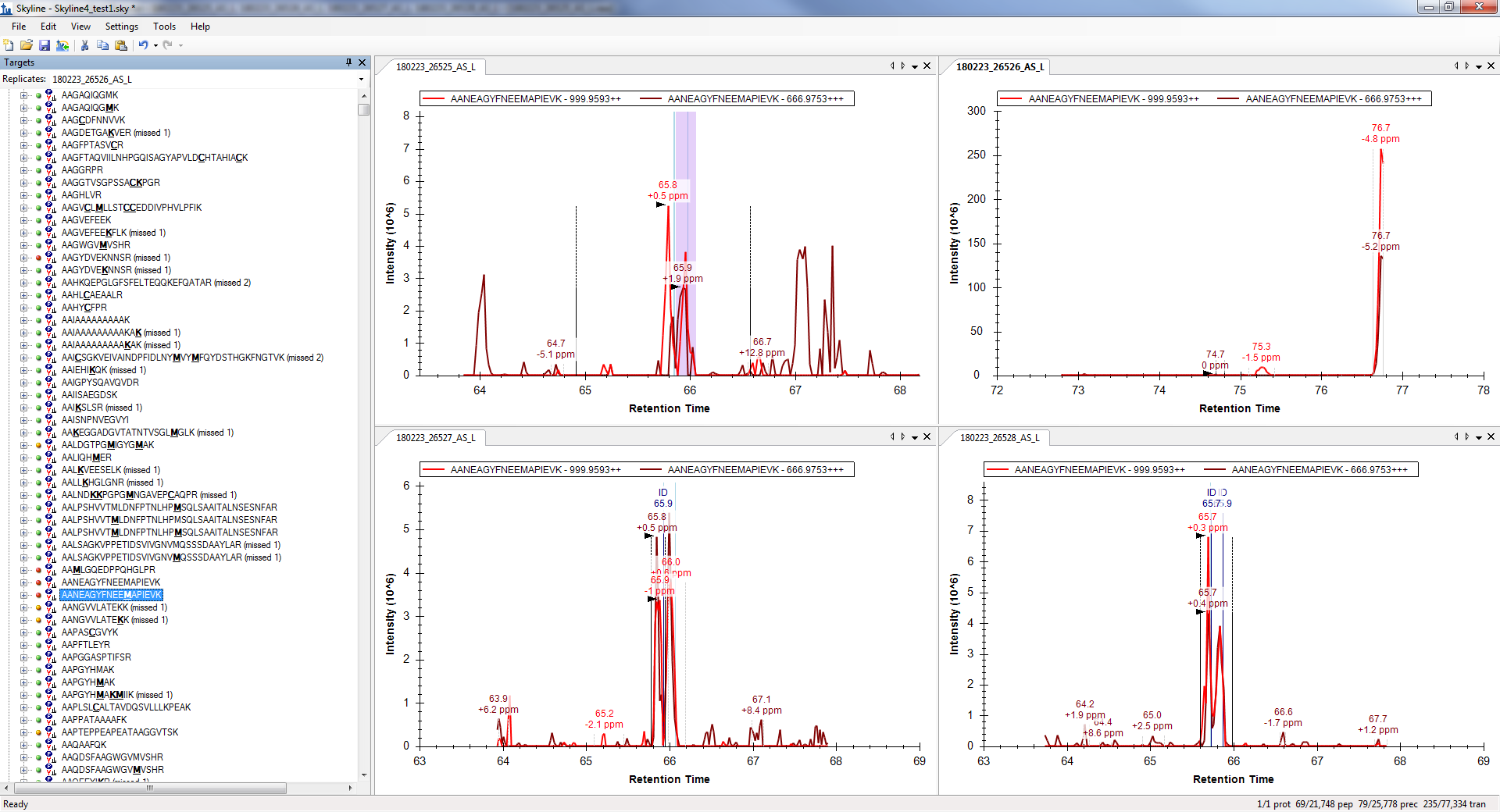 Skyline_Screenshot_AANEAG.png
Skyline_Screenshot_AANEAG.png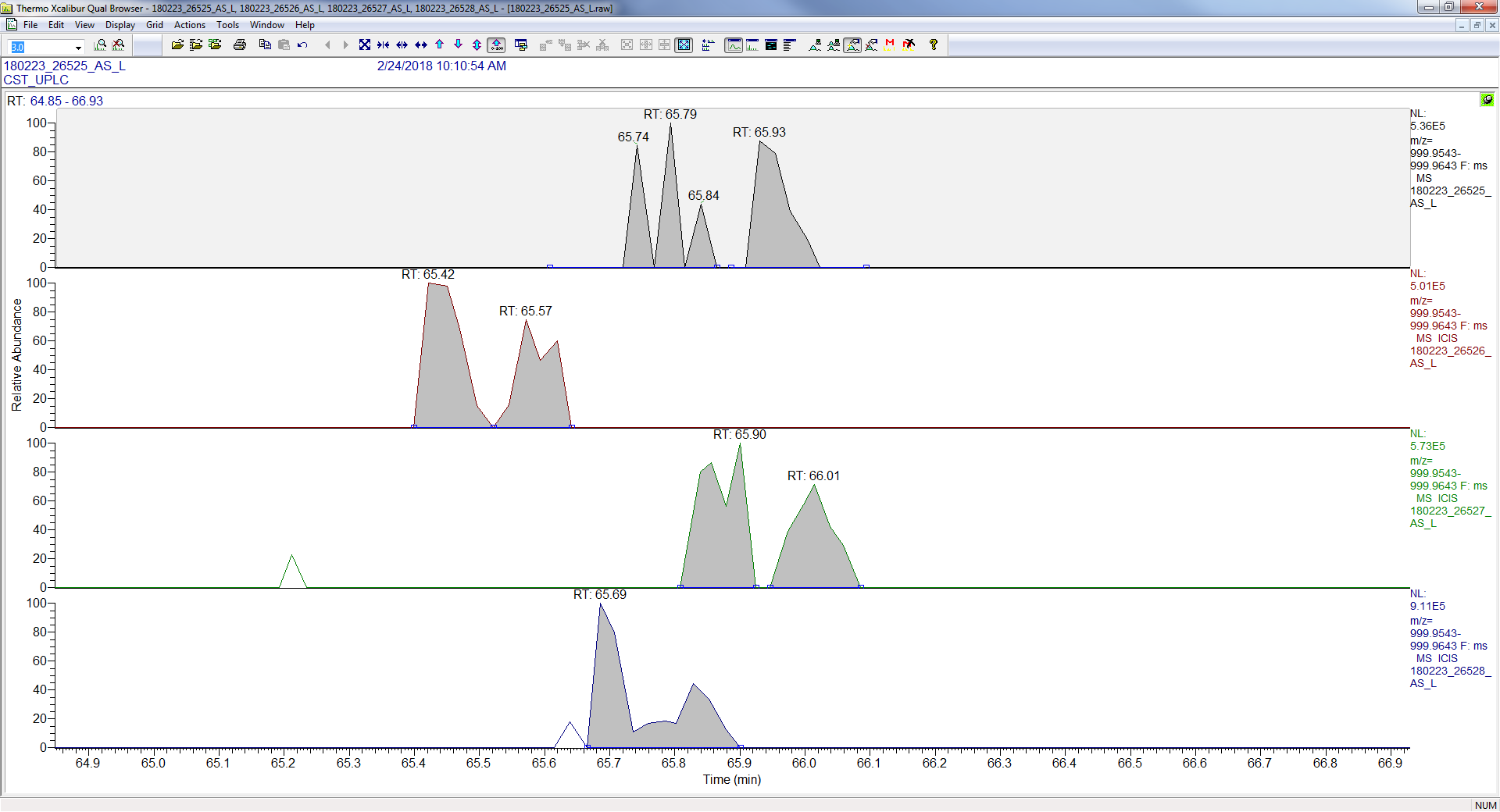 Xcalibur_Screenshot_AANEAG.png
Xcalibur_Screenshot_AANEAG.png