| |
| Nick Shulman responded: |
2018-03-15 14:24 |
That error means that the location where Skyline found your iRT peptides did not match where they were expected to be.
Sometimes this happens because the iRT peptides themselves are difficult to identify in your samples, and there happened to be so much noise that Skyline thought it found them in the wrong place.
When you get this error, Skyline does actually extract your iRT chromatograms for you, so you can look at them and see where Skyline thought that it found the peptides. Skyline just does not extract chromatograms for the rest of your peptides.
Sometimes this error happens because there are a couple of iRT standards that do not work well. That is, each one of your replicates maybe has a problem with one or two of your iRT standards, but Skyline is allowed to ignore up to 20% of the iRT standards when trying to do the linear regression. Then, in some other replicate, there is one more iRT standard that was found in the wrong place, and that linear regression fails. In that case, you would want to remove the problematic iRT standards from your list of standards in your iRT Calculator Definition.
If you send us your data, we could take a look.
In Skyline, you can use the menu item:
File > Share > (complete)
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms and iRT database.
You can upload that .zip file here:
https://skyline.ms/files.url
It would also be helpful if you gave us your .wiff file (and .wiff.scan) for your problematic replicate, and maybe one of your other replicates as well. |
| |
| brlawson responded: |
2018-03-15 15:36 |
Thanks, Nick. I was able to upload my Skyline document as well as the .wiff file for one of the files that failed, but I'm having issues getting the .wiff.scan or an addition good file to load. |
| |
| Nick Shulman responded: |
2018-03-15 16:12 |
You actually uploaded the .sky.view instead of the .sky.zip
Can you upload the .sky.zip that came from "File > Share" (or if that file is less than 50MB, you can attach it to this support request) |
| |
| brlawson responded: |
2018-03-16 09:45 |
Nick,
Sorry about the mistake. I accidentally uploaded the wrong file. The correct compressed file should be there now along with the the raw files E6 and E8. E6 imported successful whereas E8 was one of the files that failed.
Thanks again! |
| |
| Nick Shulman responded: |
2018-03-16 12:32 |
Thank you for sending your files.
Normally, the way that you figure out why the linear regression failed is by using the Retention Times Regression window, which you bring up with the menu item:
View > Retention Times > Score To Run
Then, you have to change a lot of options on the linear regression graph so that you can use it to figure out what went wrong with your iRT standards.
Choose the following things from the linear regression graph right-click menu:
Points > Standards
Replicates > Single
Calculator > iRT_PCM_test
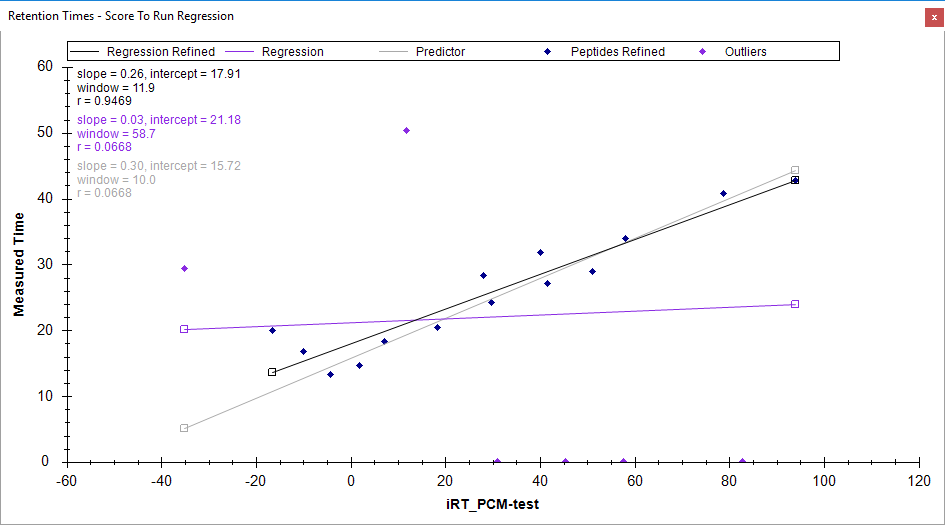
Then, in the main Skyline window you can switch which replicate is selected (it's the dropdown above the Targets tree), and you can see when you select the DataSET19_E8 replicate, it looks like the attached picture. The purple diamonds are the outliers, and if you click on them Skyline will select the peptide.
Anyway, the reason that Skyline is having so much trouble finding your iRT peptides is that you have not told Skyline which transitions are important. You are telling Skyline to look for all of the b and y ions, and Skyline has no way of knowing which of those are actually detectable, and so Skyline often chooses the wrong peak.
The attached "Sciex.sky" file has the official set of transitions that Skyline recommends for the SCIEX PepCalMix. You can import these peptides and their transitions into your document with the menu item:
File > Import > Document
(Skyline probably also offered to insert these peptides into your document when you first set up your iRT calculator) |
|
| |
| brlawson responded: |
2018-03-22 09:53 |
Nick, thanks so much for your help. In order to use the Sciex pepcal document will I need to reimport my data files using this document instead of the one that I had originally used with my imported files? Or is there a way to remove my document and add this one instead? I'm fairly new to skyline and just want to make sure I proceed correctly.
Thanks again! |
| |
| Nick Shulman responded: |
2018-03-22 12:59 |
Yes, you will need to reimport after you make those changes to the set of transitions that you are monitoring for your iRT peptides.
Whenever you change your set of peptides or transitions, you need to do a reimport.
You can reimport by going to:
Edit > Manage Results > Reimport |
| |
| sunrui responded: |
2019-03-21 10:18 |
Hi Nick,
I confused with the steps. I try to figure out what went wrong with my iRT standards. If my iRT is too less. and I can't deal with this problem by Sciex.sky |
| |
| Nick Shulman responded: |
2019-03-21 10:39 |
Hi, Sunrui,
I do not understand your question.
It might help if you could send us your files.
In Skyline, you can use the menu item:
File > Share > (complete)
to create a .zip file containing your Skyline document and supporting files including extracted chromatograms.
If that .zip file is less than 50MB then you can attach it to this support request.
Otherwise, you can upload it here:
https://skyline.ms/files.url
-- Nick |
| |
| sunrui responded: |
2019-03-21 18:19 |
Hi Nick,
I upload my file on to here:
https://skyline.ms/files.url
the name of my zip file: sr20190322_importfailed
There are 4 files, except the two pool samples. other two files are the sample, the raw file end with "t47d_2" imported fail for retention time predictor linear regression failed. another file end with "nc" imported succeed. The two files used the same ms method and the peptide concentration is also same. I wonder why this two samples have obvious differences.
Thx
Rui |
| |
| Nick Shulman responded: |
2019-03-22 03:19 |
Hi, Sunrui,
The files that you sent me did not include your spectral library ("QE_NCI60.blib") which contains your iRT values, so I cannot be sure what is going wrong.
You should always use the Skyline menu item "File > Share" when you want to send someone your Skyline document, because that menu item will make sure that the .zip file contains the necessary .blib files.
In your file "A20190321sunr_BRP_siLDHA_PRM_t47d_2.raw" it looks like you did not collect MS2 scans for a long enough time for your last two iRT peptides ("GTFIIDPAAVIR" and "LFLQFGAQGSPFLK") to be found. In that .raw file, you collected MS1 scans from 4 minutes until 22 minutes, but the MS2 scans only went from 4 minutes to 18 minutes.
In the attached picture "GTFIIDPGGVIR.png", I can see that the peptide was found in all four of your runs. However, in your "15min_pool" run, there is another 4 minutes of chromatography available after where that peptide was found, but in the other three runs, the chromatography is almost ended.
For this reason, in the other two screenshots, the peptide was only correctly identified in the 15min_pool run. In the others, Skyline seems to have chosen some other random data for the peak, because the chromatography did not extend far enough for the peptide to actually elute.
If you have an iRT predictor and Skyline is extracting chromatograms, then Skyline extracts the chromatograms in two passes.
In the first pass, Skyline extracts chromatograms for only your iRT standards. Then, Skyline attempts to do a linear regression between the time at which the peaks were found, and the iRT values in the iRT database. Skyline is trying to determine what slope and intercept will map between iRT score and observed retention time.
When Skyline is doing this regression, Skyline insists on getting an R-squared which is at least 0.99.
Skyline is willing to ignore up to two of your iRT standards in order to achieve this 0.99 threshold. If it is not possible to get a 0.99 R-squared, then Skyline refuses to extract chromatograms for the rest of your peptides.
I am actually not sure why Skyline was not able to achieve that 0.99 R-squared on your computer, since it looks like there are only two peptides that have a problem. I think the iRT values in your "QE_NCI60.blib" might be slightly different than what I expect them to be.
Hope this helps,
-- Nick |
|
| |
| sunrui responded: |
2019-03-23 01:09 |
Hi Nick,
Sorry for that I did not give you the correct form of skyline. I re-uploaded a new file, please help me to check it. the same file name with the last, but the date is recent.
https://skyline.ms/files.url
the name of my zip file: BRP_PRM_4prot_NCI60_example_20190323
And the another thing is that the same sample run another LC-MS/MS shoot 5 min longer gradient than before is okay, so in the new file I have import successfully.
Thanks
Rui |
| |
| sunrui responded: |
2019-03-23 07:58 |
Hi Nick,
It is me again. I have the problem is that whether I can change the 0.99 R-squared threshold on my own.
or if I can delete the two peptides that have a problem by myself and improve the R-squared.
Best wishes
Rui |
| |
| Nick Shulman responded: |
2019-03-24 20:47 |
Hi, Rui,
You should go to:
Settings > Peptide Settings > Prediction > Calculator Button
and push the "Choose Standards" button and remove the last two peptides.
Skyline will make you create a new .irtdb file with this new set of peptides in it, but after that you will be able to import from these .raw files without error.
-- Nick |
| |
|
|
 iRTRegresion.png
iRTRegresion.png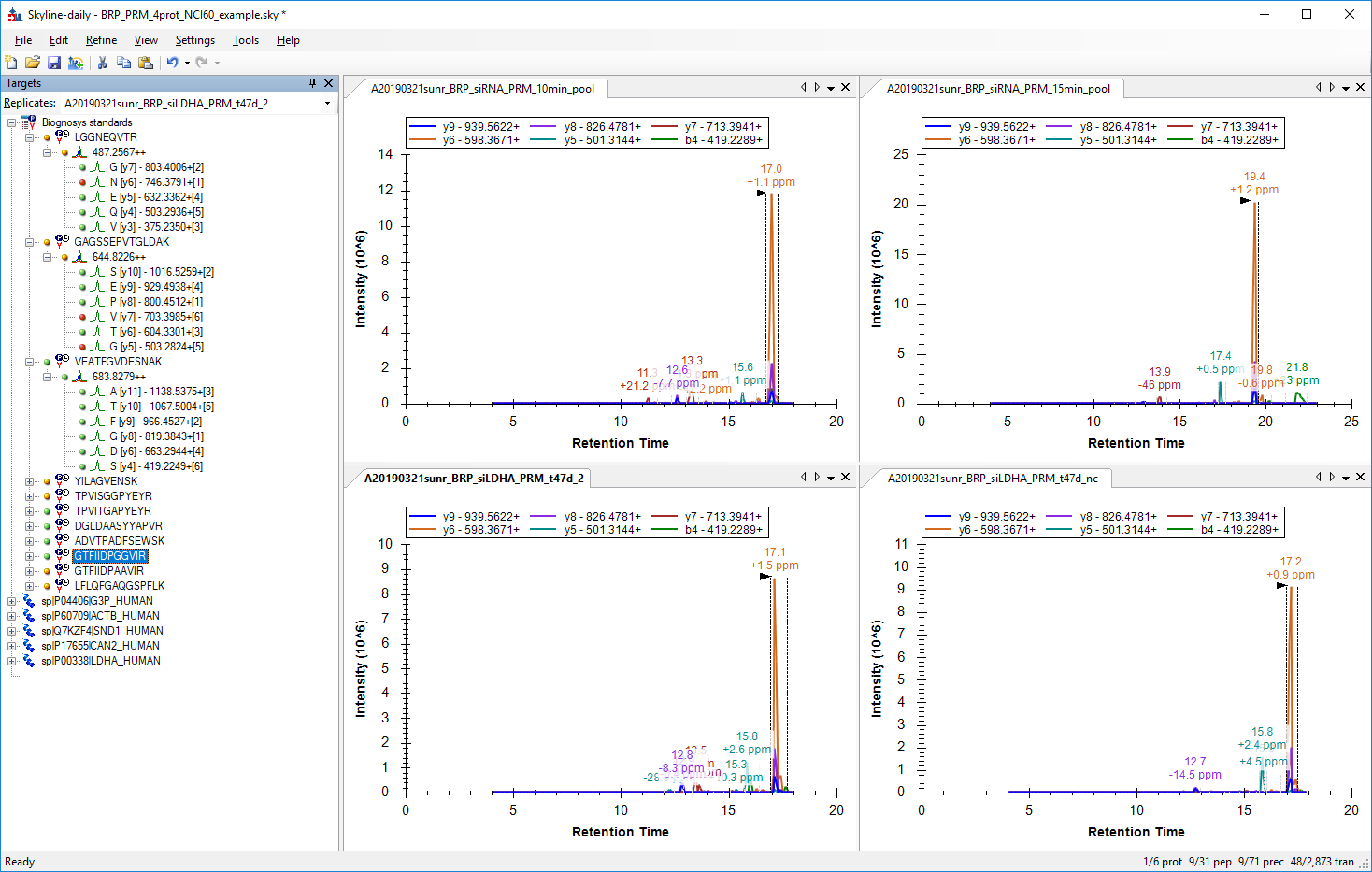 GTFIIDPGGVIR.png
GTFIIDPGGVIR.png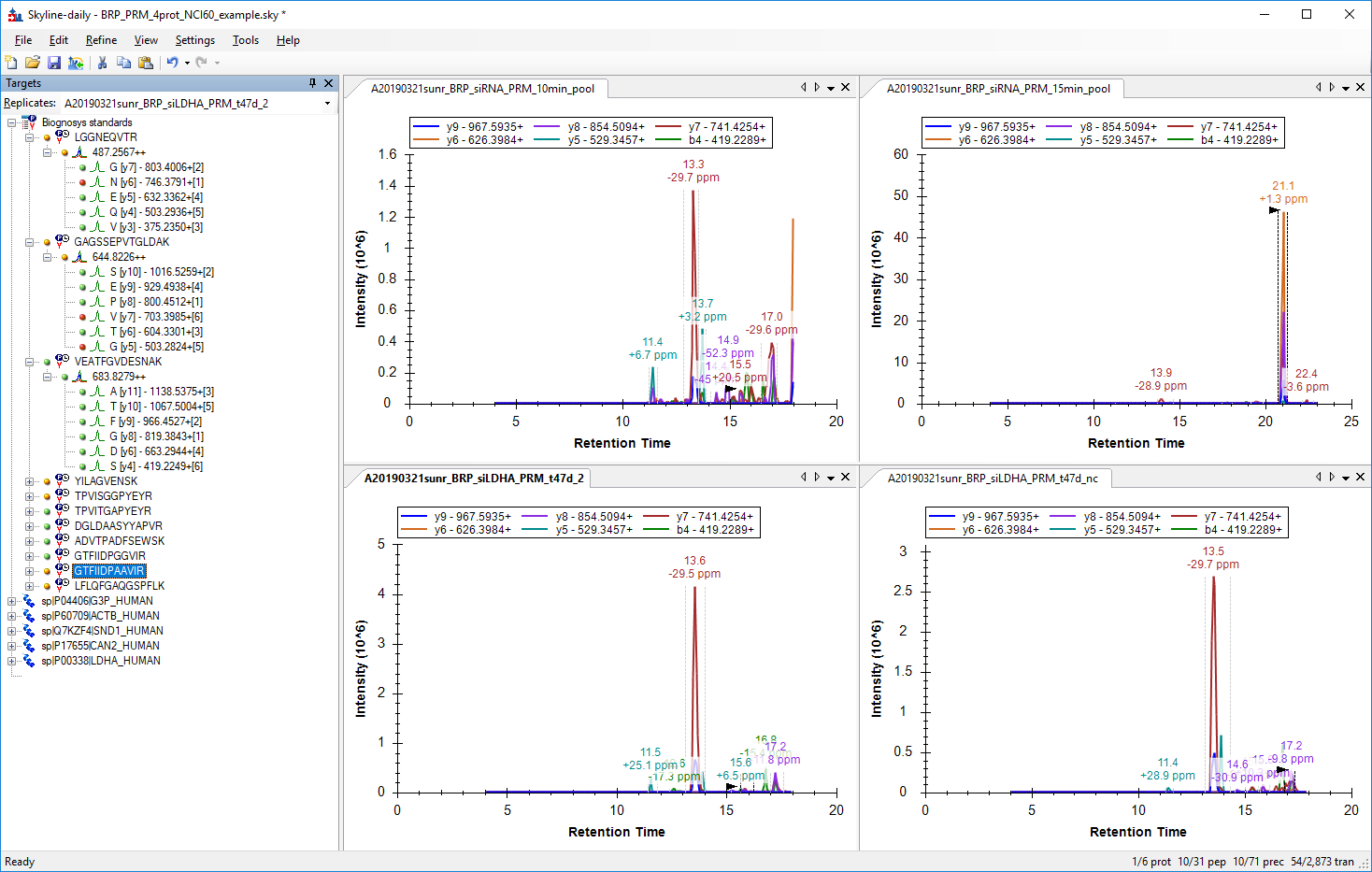 GTFIIDPAAVIR.png
GTFIIDPAAVIR.png LFLQFGAQGSPFLK.png
LFLQFGAQGSPFLK.png