Dear Skyline Team,
I am working with Skyline's MS1 Full-Scan filtering workflow to estimate the reproducibility of an O-glycopeptide enrichment protocol. In a nutshell: I have three technical replicates in which I try to verify the presence of O-glycopeptides at MS1 level using confident MS/MS identifications (both ETD and HCD).
My problems:
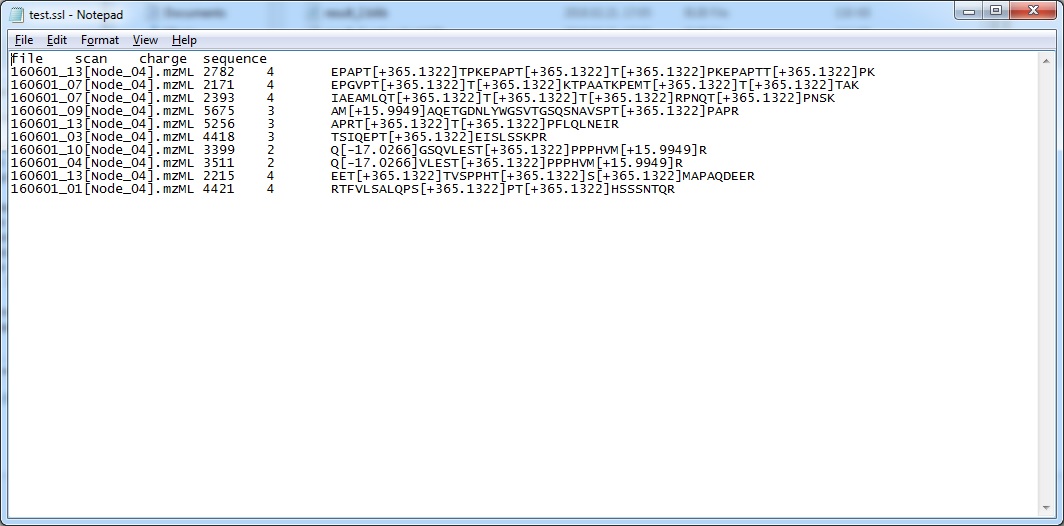
1.) I use the custom .ssl format to generate the spectral library. In this library there are ETD and HCD identifications of O-glycopeptides (for one peptide ID there is only one ETD or HCD spectrum). My problem is with the HCD glycopeptide identifications. For ETD I can insert the modifying sugar structure into the peptide sequence, but for HCD it is not acceptable because the sugar, during collisional activation, flies off from the peptide therefore the site cannot be assigned. Is there any recommendation about how to indicate the neutral loss of the sugar in the peptide sequence?
2.) I have 12 raw files in .mzML format (see raw_files.jpg). Skyline consideres for MS1 filter only the raw files that are mentioned in the .ssl file (see results_file_found.jpg and ssl_file.jpg). Why does that occur? I expected that Skyline will search the peptides, listed in the .ssl, in all the provided raw files at MS1 level.
I would really appreciate your help! Thank you!
-Adam |
| |
| Brendan MacLean responded: |
2018-02-22 01:56 |
Hi Adam,
1. Skyline supports neutral losses on modifications, or even amino acids without modification (e.g. H20 and NH3 losses). Probably your best reference for this is the Skyline Tutorial Webinar #10:
http://skyline.ms/webinar10.url
2. The wizard you are using does indeed assume that all of your raw data files have peptide-spectrum matching results. Without these, Skyline will not be able to perform retention time alignment. However, if you really want to try searching other files for your targets, you can just import those data files after you complete the wizard, through File > Import > Results.
Hope this is the help you were looking for. Good luck with your experiment.
--Brendan |
| |
| pap adam responded: |
2018-03-23 03:52 |
Dear Skyline Team,
Thank you for your kind help! I have another question about peak finding. When I did MS1 filtering it occurred frequently that Skyline was not able to find the correct peak for a given peptide in all the files provided for MS1 filtering. An example is attached where Skyline found that particular peptide in files: 7, 8, 9, 10, 12 (the files that really contain that peptide: 7, 8, 9, 10, 11, 12). So my question is: What is the cause that Skyline did not find that particular peptide in file 11. The retention time for file 11 is not OK, it is around 21 min instead of ~26.7, when the peptide is really eluting. I tested my dataset with another software and it was able to find that peptide in file 11. How peak-findig works in Skyline?
Thank you for your kind help!
-Adam |
|
| |
| Brendan MacLean responded: |
2018-03-23 08:57 |
Hi Adam,
It looks to me like you don't quite have MS1 filtering set up correctly. There are no ID annotations appearing at all, which means you are forcing Skyline to pick peaks as if the ID retention times in your spectral library come from an entirely different data set, instead of the same files from which you are extracting chromatograms.
Please have a look at the tip page for diagnosing this issue:
https://skyline.ms/wiki/home/software/Skyline/page.view?name=mascot_missing_rt
I think the most likely explanation is a name change between the files you searched to build your library and the files you are using for chromatogram extraction.
Skyline will do much better at peak picking if you can get actual ID annotations. Another indicator is what you see with View > Retention Times > Alignment. If you do not see good alignment information for all of your runs in this view, then you should be working to debug why not and not continuing with a suboptimal analysis.
Hope this helps. Let us know what you find.
--Brendan |
| |
| pap adam responded: |
2018-03-26 02:21 |
Hi Brendan,
I checked my filtering results, and I see ID annotations for every peptide. The .ssl file that Skyline used to build the spectral library contains one ID for every peptide, therefore when I filter my 12 files there will be only one file with ID annotation (see Figure_1 in the attachments, the ID annotation is in a red rectangle). Can this be the problem? I thought when there is ID annotation in one file, Skyline will use this RT value in all the remainig files to search for a particular peptide's MS1 trace. How does peak picking work? Please explain.
I checked the RT alignment as you suggested. Skyline couldn't do the alignment, because when I checked the RT alignment window (View > Retention Times > Alignment) there was no regression fit nor R square values (see Figure_2). From this I think that to make the RT alignment across files for a particular peptide there has to be ID annotations from all the 12 files. Is that correct? How does the alignment work? Please explain.
I also checked the source files for spectral library building and for chromatogram extraction. They are the very same files, with a very simple name: 1.mzML ... 12.mzML. In the .ssl these names were used (see Figure_3 and 4).
I use the .ssl file format for spectral library building because a glycopeptide IDd from HCD in ProteinProspector has the following format: PEPTIDE SEQUENCE+MODIFICATION (e.g.: IAPTVWK+365.1322). As it turned out Skyline cannot handle this sequence format.
Thank you for your kind help,
-Adam |
|
| |
| Brendan MacLean responded: |
2018-03-26 10:47 |
So, not having alignment work is the crux issue for you. The way it is supposed to work is that redundant IDs across your files allow Skyline to perform a linear regression to produce a linear equation for mapping between runs.
Since, it sounds like you have constructed already a non-redundant SSL file, there are no shared IDs between your runs, and Skyline cannot perform the regression.
This is unexpected and makes it impossible for Skyline to do retention time alignment, which means Skyline just ends up using the retention times from the other runs without alignment, and those times are given less weight in peak picking, as they should be without any alignment.
You should think about whether you can put back some redundancy into your SSL file, where you have confident assignments for a peptide in multiple files, rather than always just choosing what you feel is the best assignment across all your runs.
Hope this helps. Thanks for the screenshots.
--Brendan |
| |
|
|
 ssl_file.jpg
ssl_file.jpg raw_files.jpg
raw_files.jpg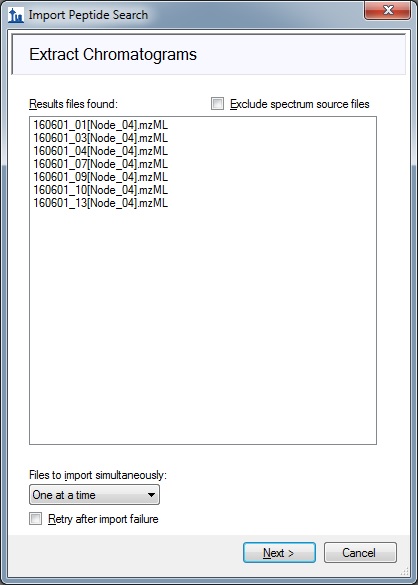 results_file_found.jpg
results_file_found.jpg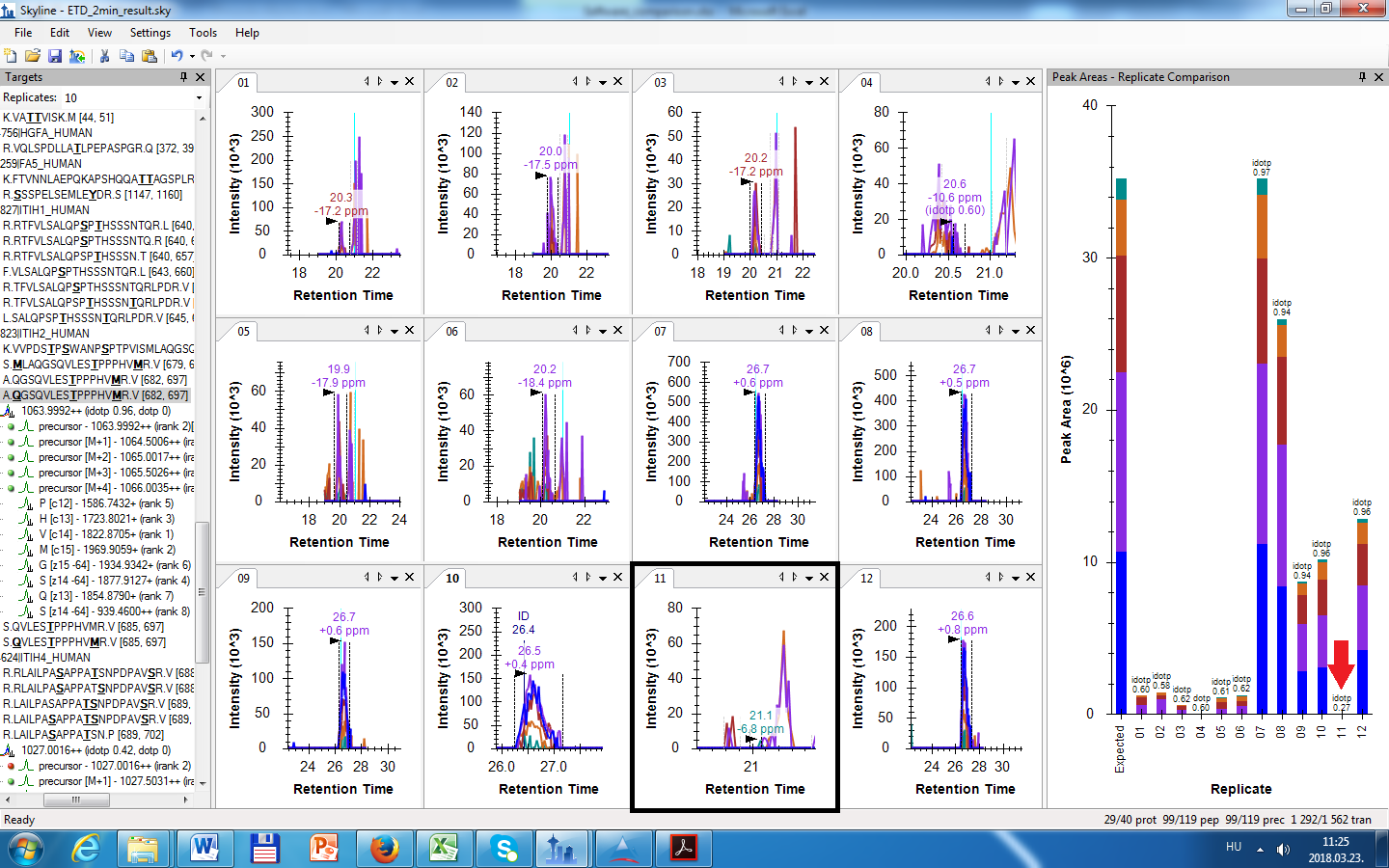 missing_peak_Skyline.png
missing_peak_Skyline.png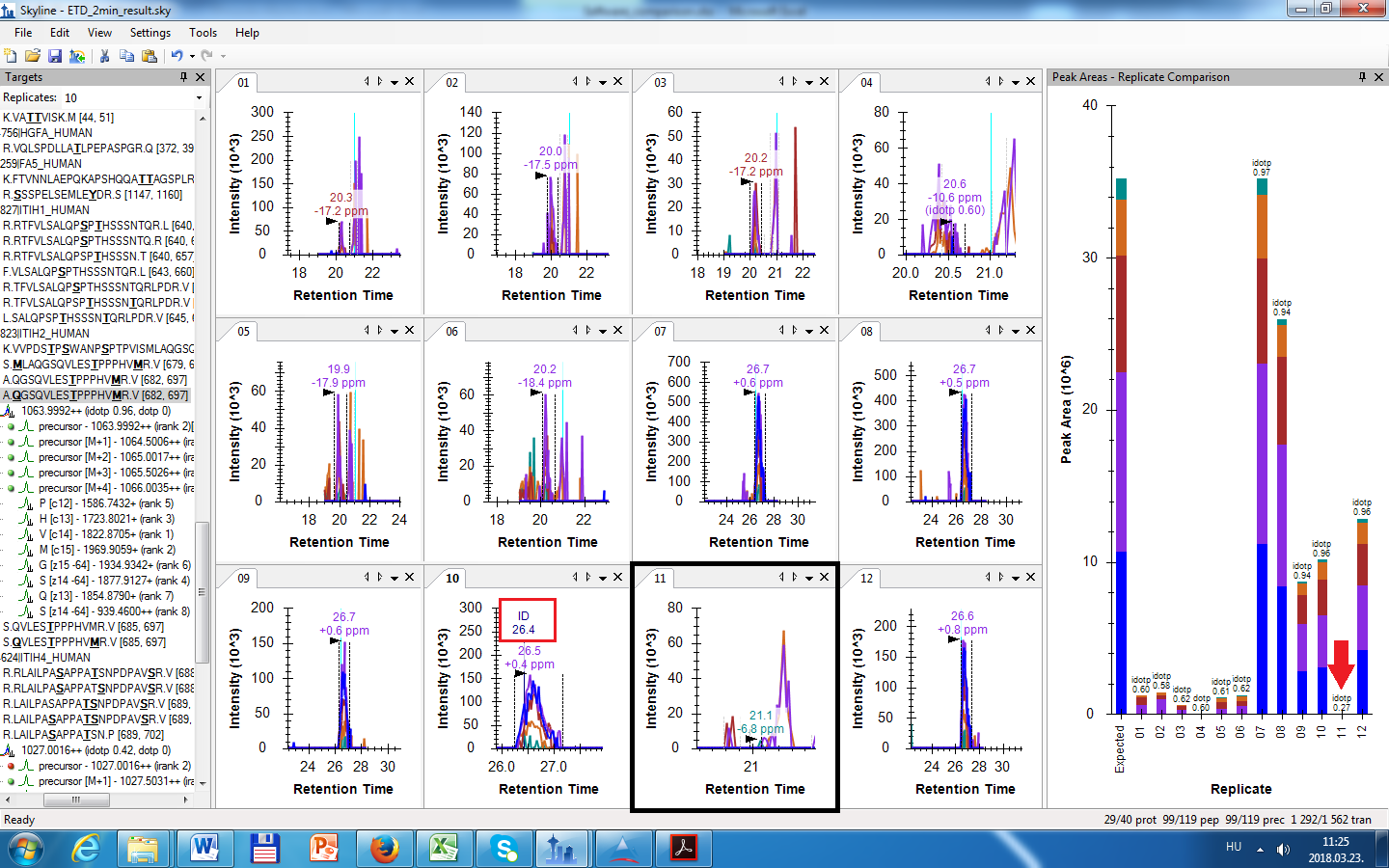 Figure_1.png
Figure_1.png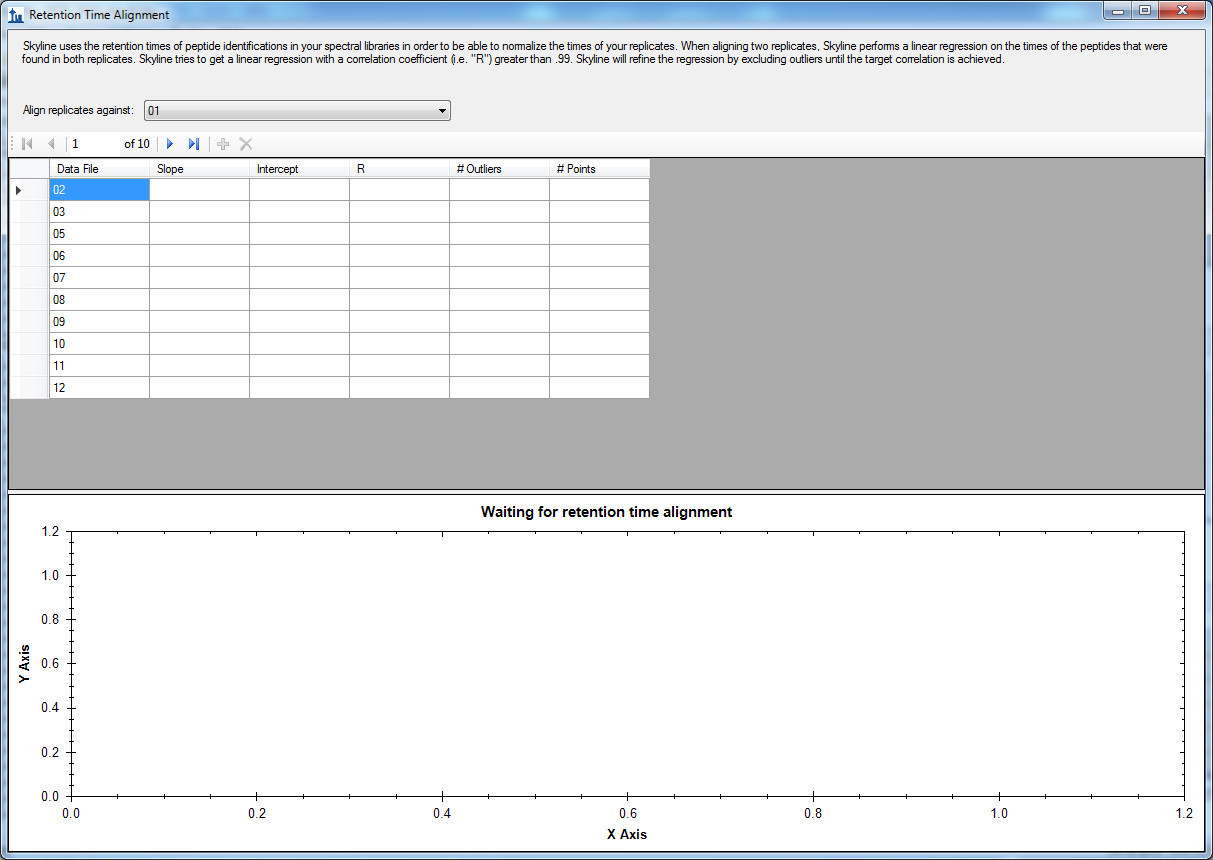 Figure_2.png
Figure_2.png Figure_3.png
Figure_3.png Figure_4.png
Figure_4.png