| targeted metabolomics with Skyline | arnaud garcia | 2018-02-08 10:44 | |||||||||||||||||||||||||||||
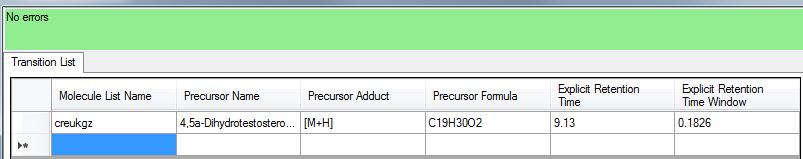
Dear Skyline developpers, I work on a project of using this software to extract feature from Orbitrap raw data to obtain a standardized profile of metabolites. The goal is - Upload raw data files - Upload a database of molecules to screen for ( with exact masses/ formula and known retention time) - Extract from the database the area of the masses that are found only in the time windows (and if there is nothing, then the area should be left blank). - Get in an excel file (in the best scenario) the observed area, peak info, masses and retention time for each molecules screened. After uploading the list (using the "Edit > insert > transition list") and the insertion of raw data, it occurs to me that I couldn't get the selective windows of retention time correctly even after "re-scoring" of the data and changing parameters. For many metabolites it was good because they were found within the right range. However (fig 1 in the file attached), many ions were returned by the software to be matching our molecules of interest while they were out of the suggested ranges. Same, another problem was the mass accuracy of my list. I was working with an Orbitrap format and couldn't reach manually the mass accuracy settings ( from Settings > Transition Settngs > Full Scan). The accuracy at my selected settings were too high for my study so i had to switch my precursor mass analyzer to "centroided" in order to manipulate the mass accuracy, even if it is less than ideal. So here i have two main questions : - is it possible to integrate areas automatically for a set m/z windows and a set Retention time windows regardless of the kind of data used (orbitrap, Waters, Agilent..)? - Is there a way to select easily the mass accuracy of the selection of masses when selecting MS1 Orbitrap files? Retention time accuracy and mass accuracy are really important for selectivity in metabolomics and since the goal of Skyline is also to cover small molecules integration, those parameters are still confusing to me. Thank you very much and have a lovely day, Arnaud |
|||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||
 Capture5.JPG
Capture5.JPG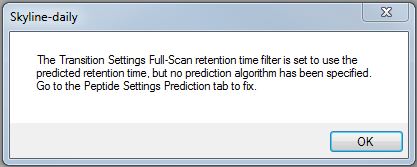 Capture2.JPG
Capture2.JPG