| |
| Nick Shulman responded: |
2017-09-19 22:52 |
Can you send us your Skyline document?
In Skyline you can use the menu item:
File>Share>(Complete)
to create a .zip file containing your Skyline document with supporting files including extracted chromatograms.
You can upload that file here:
https://skyline.ms/files.url |
| |
| k valgepea responded: |
2017-09-21 00:18 |
Thank you for the quick reply Nick!
It took 24h, but a the file you requested has now been uploaded with the name "Global-no label HighBC MS1in.sky". It includes the data with all p, y, and b-ions selected during data import.
Please let me know how you go. Good luck! |
| |
| Nick Shulman responded: |
2017-09-21 04:38 |
Thanks for sending me your files.
One thing that I noticed about your Skyline document is that you have approximately the same number of Y and B transitions. Usually Y ions are much easier to detect, and I would expect to see a lot more of them.
When I look at the matched spectrum for the first peptide in your document (see attached image), Skyline only thinks that it sees the y1 and b5 ions. There is also a beautiful spectrum for this same peptide in your "33-36" library, but the one I am showing here in the "25-32+59" library apparently got a slightly better percolator score (0 versus .009951; 0 is the better score).
One thing that I thought might be going on was your Ion Match Tolerance in:
Settings > Transition Settings > Library
You have it set to .05. If you have that set to the wrong number, then Skyline will not get the right answer when Skyline tries to decide which were the most intense ions observed in your library spectra. However, even when I change that match tolerance to 1, it does not make the spectrum look any better in terms of which ions Skyline believes were present.
If Skyline does not see the same things in your spectra as your peptide search engine saw, Skyline is going to choose the wrong transitions to extract chromatograms for. If these transitions are actually undetectable, or were not selective enough, then your targets and your decoys are going to look too much alike.
Hope this helps! |
|
| |
| Brendan MacLean responded: |
2017-09-21 06:33 |
Hi Kaspar, There really seem to be a lot of problems with this document for what you are trying to achieve.
- The normal number of recommended fragment ion transitions (from the original Gillet, MCP 2012 paper) would be 6, and you are setting 5 in the Transition Settings - Library tab.
- In the case Nick shows, there is only 1 fragment transition. If apply a minimum 8 (3 precursors + 5 fragments; normal would be 4-6) using Edit > Refine > Advanced, then the number of target precursors drops from 15,307 to 2,960. Even with a minimum of 5 (3 precursors + 2 fragments) the number of targeted precursors drops to 8,374. So, nearly 1/2 of your peptides have only 1 or 0 fragment ions. This leaves me feeling there is a bug in the Library dot-product score since it should need at least 3 fragment transitions to be calculated. It should be showing up gray and unusable.
- The Product mass error score is showing up gray and unusable, which indicates some precursors have no fragment ions at all.
- There is no retention time predictor in Peptide Settings - Prediction tab. You are using the previously measured DDA library times to predict the times in DIA. This is the weakest type of retention time prediction, and we would expect much better results with an iRT predictor, which would allow for run-to-run alignment.
When I apply a minimum of 5 fragment ions (though I would prefer to see 6) - leaving only 2,960 targeted precursors. Then at least I get a reasonable looking model with some separation of good peaks from decoys (see attached). If as Nick suggests, I widen the Ion match tolerance in the Transition Settings - Library tab to 0.5, then I get 6,656 precursors with 5 fragment ions, but I can't train on this, because decoys would need to be regenerated and all of the data reimported. In fact, I get the same number if I use the 6 fragments suggested in the SWATH paper. What is the source of your library spectra? It doesn't seem like they are from the same high-accuracy detector as the DIA MS/MS are using. A match tolerance of 0.05 would normally work well for spectra acquired on an Orbitrap. So, final suggestions:
- Delete your decoys.
- In Transition Settings - Library tab change Ion match tolerance to 0.5 and Pick 6 product ions.
- Edit > Refine > Advanced with Min peptides per protein of 1 and Min transitions per precursor of 8.
- Edit > Refine > Add decoys.
- Edit > Manage Results and reimport all of the data.
This should give you something that will produce a usable peak picking model. You should also consider using iRT for retention time prediction, as this will produce a better model and better peak picking. Hope this helps. Good luck. Thanks for posting the data. It took us less than 1 minute to download and we couldn't possibly have worked this out without it. --Brendan |
|
| |
| k valgepea responded: |
2017-09-21 18:57 |
Thank you very much for the detailed replies and your time investment in looking into my data! I will try what you recommended but I am puzzled why the ion match tolerance of 0.05 gives such bad results. The library files were generated by ProteomeDiscoverer using the same MS2 0.05 tolerance as both DDA and DIA were run on the same HF Orbitrap. Two questions regarding only using MS1 data:
1) I cannot find where I could specify the precursor mass tolerance? Or its irrelevant?
2) Would the Skyline default or mProphet work with only precursor data? Assuming I import 5 or 6 precursor ions per precursor. Thank a lot again! |
| |
| Nick Shulman responded: |
2017-09-21 19:46 |
The mass tolerance that I pointed you at on:
Settings > Transition Settings > Library
only affects the way that Skyline interprets spectra in a library for the purpose of deciding which transitions should be added to your document. When you ask about precursor mass tolerance, it sounds like you might be talking about chromatogram extraction width. That is controlled by the resolution settings on:
Settings > Transition Settings > Full Scan
and it's explained in the MS1 full scan filtering tutorial (page 10):
https://skyline.ms/_webdav/home/software/Skyline/@files/tutorials/MS1Filtering-1_2.pdfIt might be worth trying to figure out why the spectrum in my screenshot which clearly does not look like the peptide that it's supposed to got such a good score in your library. Reasons that I could think of might be that BiblioSpec is misinterpreting the zero score that it was assigned, and in this case zero is supposed to be a very bad score. Or it might be that that's not actually the spectrum that your peptide search engine was looking at, and that either BiblioSpec or your search engine has some sort of off by one error in terms of referring to spectra in the mass spec data file. I don't think it makes sense to talk about having 5 precursor ions. The different precursor ions come from the different masses in the isotope distribution, and, once you start looking at the M+4 mass, the expected intensity is so low it's not worth measuring. |
| |
| Brendan MacLean responded: |
2017-09-21 20:19 |
Why would you want to try precursor only peak matching when you are acquiring DIA data? That would be a huge waste of instrument time. Most of your time was devoted to collecting MS/MS spectra. You should be using them. If you want to try MS1 only, then you would typically want to devote as much instrument time to that as possible. But, yeah, I would agree with Nick. The MS/MS spectra look unbelievably poorly matched, and if they really are supposed to have <50ppm mass accuracy (which 0.05 is for 1000 m/z), then it is hard to believe these spectrum matches, and you should look into your search pipeline, and ideally a tool that can show you the peptides and spectra that were matched at what confidence level, and then you can compare those to what is showing up in your library. |
| |
| Brendan MacLean responded: |
2017-09-21 20:23 |
You can use View > Spectral Libraries to see what Skyline has put in your libraries. They are not completely free of believable matches, but there are plenty that are hard to believe. I think you will have to agree, no matter what ion match tolerance you use. Just take a few of those and track them back into your peptide search software to try to understand whether you believe the right peptides and spectra are ending up in your library. |
| |
| k valgepea responded: |
2017-09-21 23:43 |
Thanks a lot again for your input! I re-imported the data you used with 0.5 tolerance, picked 6 ions (+ still imported 3 peaks for precursors), filtered for min 8 transitions per precursor and 2 peptides/protein (I am not sure about quantification based on 1 peptide). Now the mProphet looks reasonable but I only have 720 proteins to work with compared to the 1875 in the library… I am not sure though why did this result in 3781 precursors, compared to the 6656 you saw... I have thought that (Thermo) people like to use MS1 data from Thermo HF SWATH runs as the Orbitrap gives you superior sensitivity compared to other detectors and that MS1 signals are much more intense..? Thanks again! |
|
| |
| Brendan MacLean responded: |
2017-09-22 04:41 |
This is a common misconception I try to teach about when I get the chance. Sensitivity in mass spec is not solely based on the number of ions that reach the detector. It is based on signal-to-noise. Although signal is reduced by introducing a new stage of analysis in MS/MS, noise may be reduced even more (depending on the level of filtering at Q1), and therefore, sensitivity may increase despite the fact that fewer ions reach the detector. This effect does depend somewhat on the complexity of your sample, but in all but highly purified samples, we see improved dynamic range with MS/MS chromatograms even at 25 m/z isolation windows in SWATH. Narrowing those windows further improves the selectivity and the sensitivity to the point that you are able to maintain enough points-across-the-peak to reconstruct the ion elution accurately. Again, this is despite the fact that narrowing Q1 obviously reduces the number of ions that reach the detector, but the increase in selectivity appears to be worth that. This is why you see a transition in the field from the original 32 fixed windows described in the Gillet, MCP 2012 paper to 64 variable width windows. A transition which is justified in the Navarro, Nature Biotech 2016 paper. As for your library, please follow our advice to dig deeper into your expectations and what is actually happening with your peptide spectrum matching. If the sense of your probability values is reversed, then simply setting a 0.05 cut-off will not help, since Skyline is already using 1-probability in applying the cut-off. This means that if, counter to expectation, 1 turns out to be good in your native scores then a 0.95 cut-off will exclude all matches below 5% FDR, and a 0.05 cut-off would exclude all matches below 95% FDR. Maybe you should post ( http://skyline.ms/files.url) for us the files you are using to build your library. So we can have a closer look ourselves at what may be going on. It really doesn't sound like you are getting a valid library. --Brendan |
|
| |
| k valgepea responded: |
2017-09-26 22:18 |
Thank you Brendan for the reply! I completely agree with you on MS2 quantification being superior, but that is what I concluded to be the probably reason for HF DIA data being quantified at MS1 level in Thermo posters. I have confirmed that the Percolator scores are not reversed: lower means higher confidence. A question regarding you earlier comment on using iRT predictor instead of DDA library times for DIA peak picking: would there be instructions somewhere how to calibrate the iRT predictor for DIA data using some of the target peptides (not iRT spike-ins)? I was scanning through the support board without luck and I wasn't smart enough to figure this out based on the iRT tutorial... Thanks again! |
| |
| k valgepea responded: |
2017-10-05 03:19 |
Hi again guys! I am just letting you know that indeed the issue was with the library files. In the file I sent you, I used two library files (ticked two in the Library box) and evidently Skyline had a problem with that. I now get good results with using one library file (essentially the two combined in one pre-Skyline), and actually get acceptable peak picking even when using either one of the separate files. Thanks again! |
| |
|
|
 p values (precursors+fragment ions).png
p values (precursors+fragment ions).png p values (precursors).png
p values (precursors).png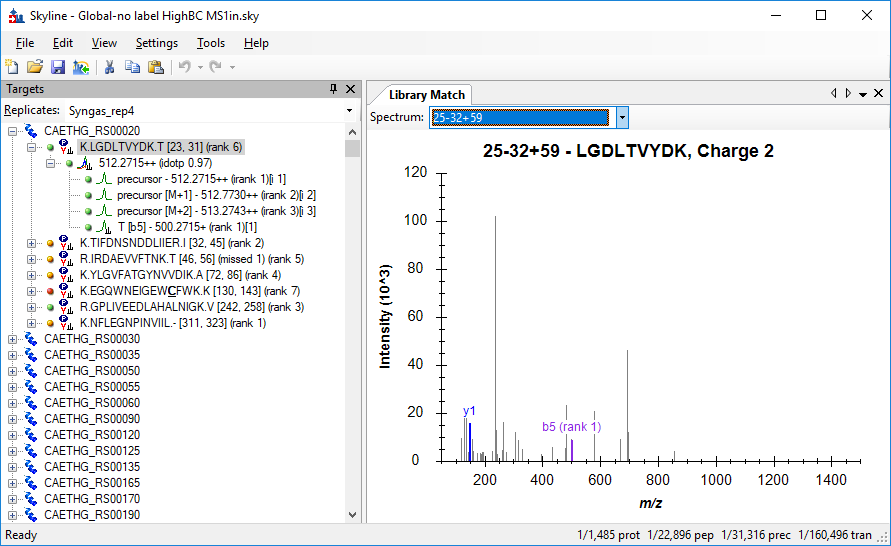 LibraryMatch.png
LibraryMatch.png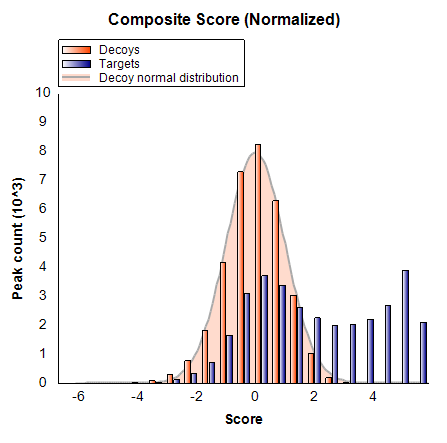 Model with 5 fragments.png
Model with 5 fragments.png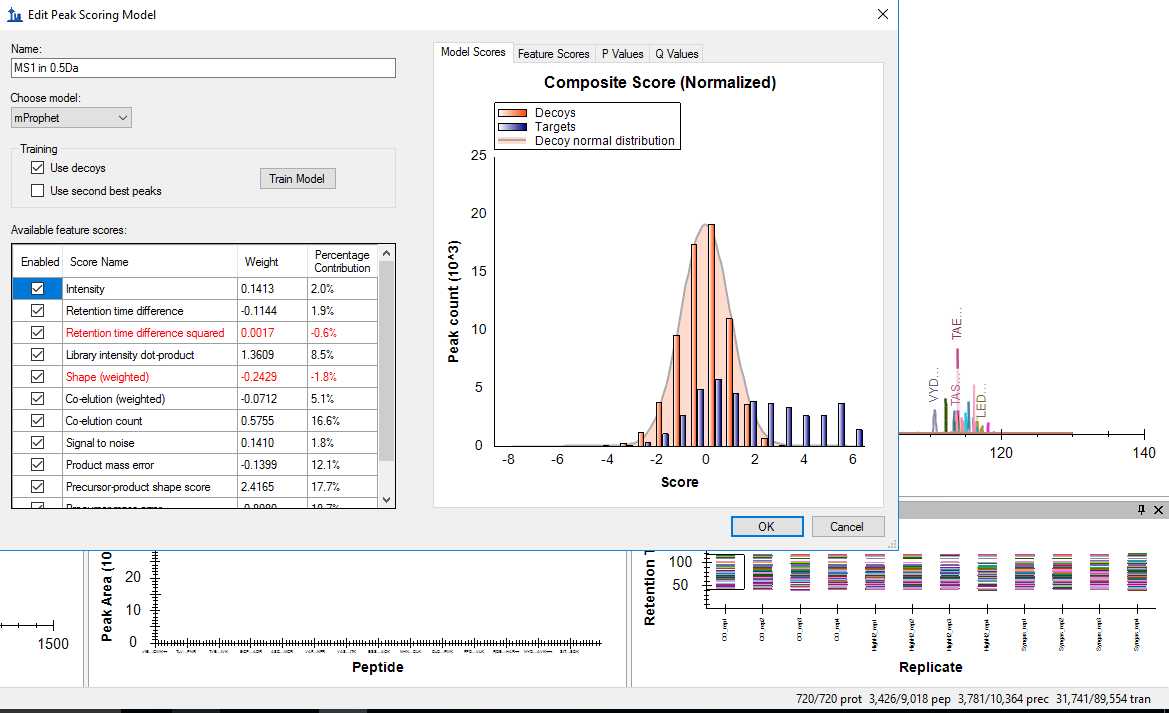 mProphet peak picking.png
mProphet peak picking.png