Dear,
If I use different RT with different mass range, how should I set my isolation window. That means I use 300-1000 M/Z in a period of 20 mins, 500-1200 M/Z in another period of 20 mins. The "results only" can work for my data? look forward to your reply. |
| |
| Brendan MacLean responded: |
2017-08-07 06:43 |
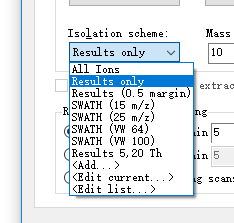
It depends on whether you use the classic SWATH overlap in measured isolation windows of 1 m/z and want 0.5 m/z margins excluded from your extraction windows, as described in the original Gillet, MCP 2012 SWATH paper.
If you do, then you would want the "Results (0.5 margins)" option.
The "Results" option will be best when you are using "Optimized window placement" without excluding any margins from extraction, as described in the Eggertson, Nature Methods 2013 MSX paper.
Finally, if the isolation windows simply cover a different range, but are either included or excluded based on time, then you can also use a list of ranges specified explicitly. That this, the following would be fine:
300-401
400-501
500-601
600-701
700-801
800-901
900-1001
1000-1101
1100-1201
It would not matter to Skyline in this case that you are not measuring all of your windows over the entire gradient. As long as you have a fixed set of windows to cover the range, Skyline will extract chromatogram points from the right scans.
If, however, you significantly change your measurement scheme between the two time ranges, then you are indeed limited to a Results-based solution. Here is an example:
0-20 minutes:
300-401
400-601
600-801
800-1001
and
20-40 minutes:
500-701
700-901
901-1200
If these were your true isolation scheme, Skyline could only deal with this using either Results or Results with a margin.
Thanks for posting your question to the Skyline support board.
--Brendan |
| |
| chia-lin responded: |
2017-08-08 18:09 |
Dear Brendan
Thanks for your reply. I meet another problem.My sample are endogenous peptides, so I don't need choose any digestion enzyme.How can I set here? I am looking forward to your reply. |
|
| |
| Brendan MacLean responded: |
2017-08-08 21:45 |
You can just leave the Enzyme setting on Trypsin. There is not Enzyme setting that would allow you to "digest" proteins to meet your requirements. Instead, with Skyline, you will need to pursue a strategy of getting targets into your document that does not involve insilico digestion. You have the following options:
- Just paste a line separated list of peptides directly into the Targets view or Edit > Insert > Peptides. These can be any peptides which need not be achieved through any particular cleavage of proteins. You can group them however you like with names that match proteins, or groups of proteins, or any explanation you like.
- If the peptides are contained in a spectral library that resulted from a peptide-spectrum-matching pipeline (e.g. Mascot, Protein Pilot, Proteome Discoverer, MaxQuant, etc.), then you can use the Spectral Library Explorer from View > Spectral Libraries and click the Add or Add All button to add peptides to your document wherever you place the selection. If the selection is in a peptide list, then new peptides will be added to that list. Otherwise, a new list named "Library Peptides" will be created, but you can easily rename this list by selecting it and typing a new name.
- If you still want your peptides added correctly associated with their proteins of origin, then you need to build a Background Proteome (which is described in the Targeted Method Editing tutorial - http://skyline.ms/tutorial_method_edit.url). Once you have one set-up then the Add or Add All buttons in the Library Explorer will also support "Associate proteins" being checked.
Many ways to get non-enzymatic cleaved peptides into your Targets list, but you can't just paste a protein sequence or import a FASTA file or use Edit > Insert > Proteins or FASTA. Those features require enzymatic cleavage. Though, as you probably noticed in the screenshot you posted, we added support for semi-enzymatic cleavage in version 3.7, and a default example "Trypsin (semi) P". It is possible to define other semi-enzymatic cleavage. Hope this helps. Thanks for using Skyline in your research. --Brendan |
| |
| chia-lin responded: |
2017-08-09 19:20 |
thank you. I have tried as you say and it worked well. |
| |
|
|
 微信图片_20170807151841.png
微信图片_20170807151841.png.jpg) 1502240576(1).jpg
1502240576(1).jpg