| Peak boundaries for small molecule shotgun lipidomics | bharat | 2017-07-19 13:49 | |||||||||||||||||||||||||||||||||||||||||||||||||
Hello: I am interested in using skyline instead of Analyst for processing data from shotgun lipidomics. These runs are generated without a column, so all we're interested is an area under the curve from a start time to an end time. After these raw areas are generated, all other work on deconvolution, quantitation, etc are done externally. So, all we would need is for the integration to be done somewhat robotically from start time t1 to end time t2. I've been very successful, thanks to the good documentation, in getting all the transitions in (>140) and the integrations done. But, I can' get to the final step, which is this automatic peak boundary picking. So, you can see from the attachment that sometimes we get the good, and sometimes the needs adjustment. I've tried the "import peak boundaries" option, but that doesn't seem to work for me for small molecules. Anything I'm missing here? Thanks Bharat |
|||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
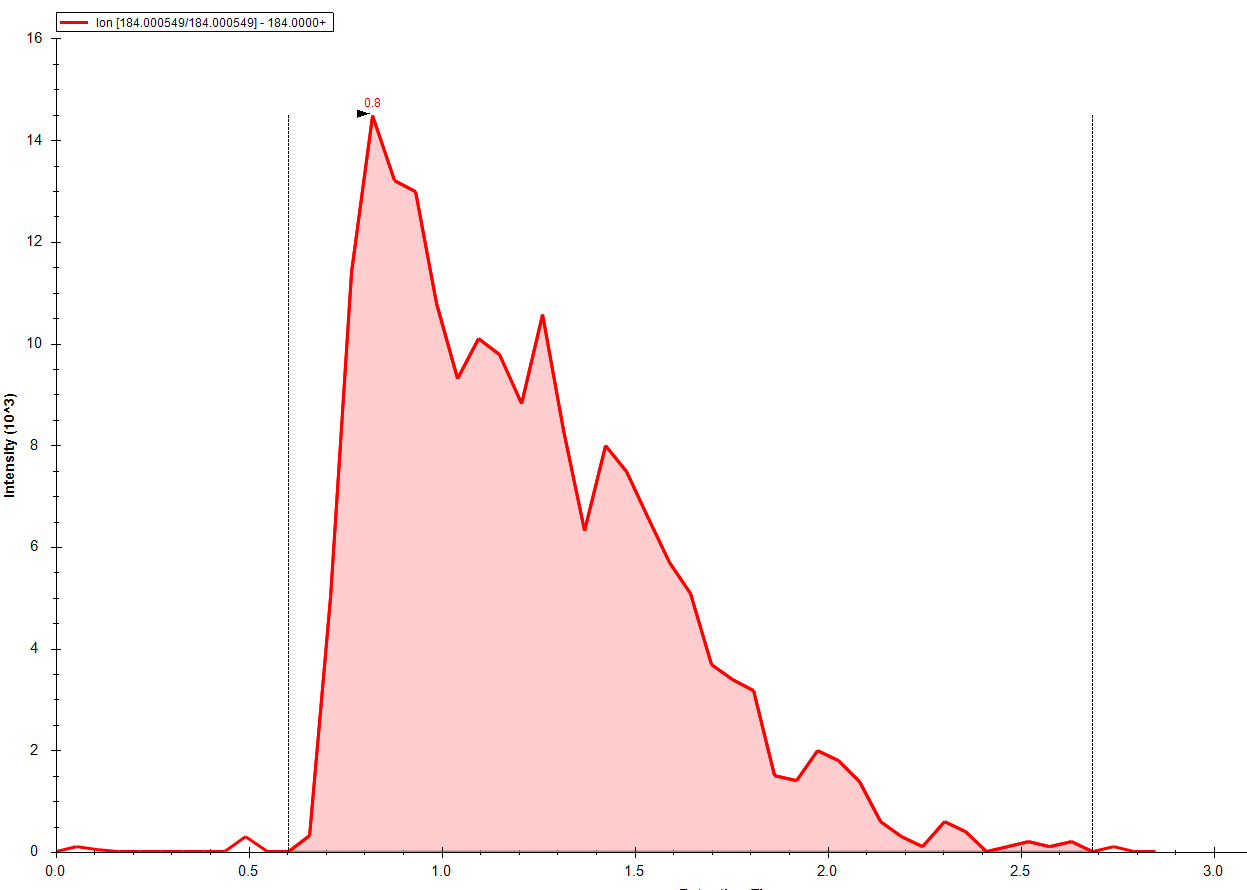 GoodIntegration.PNG
GoodIntegration.PNG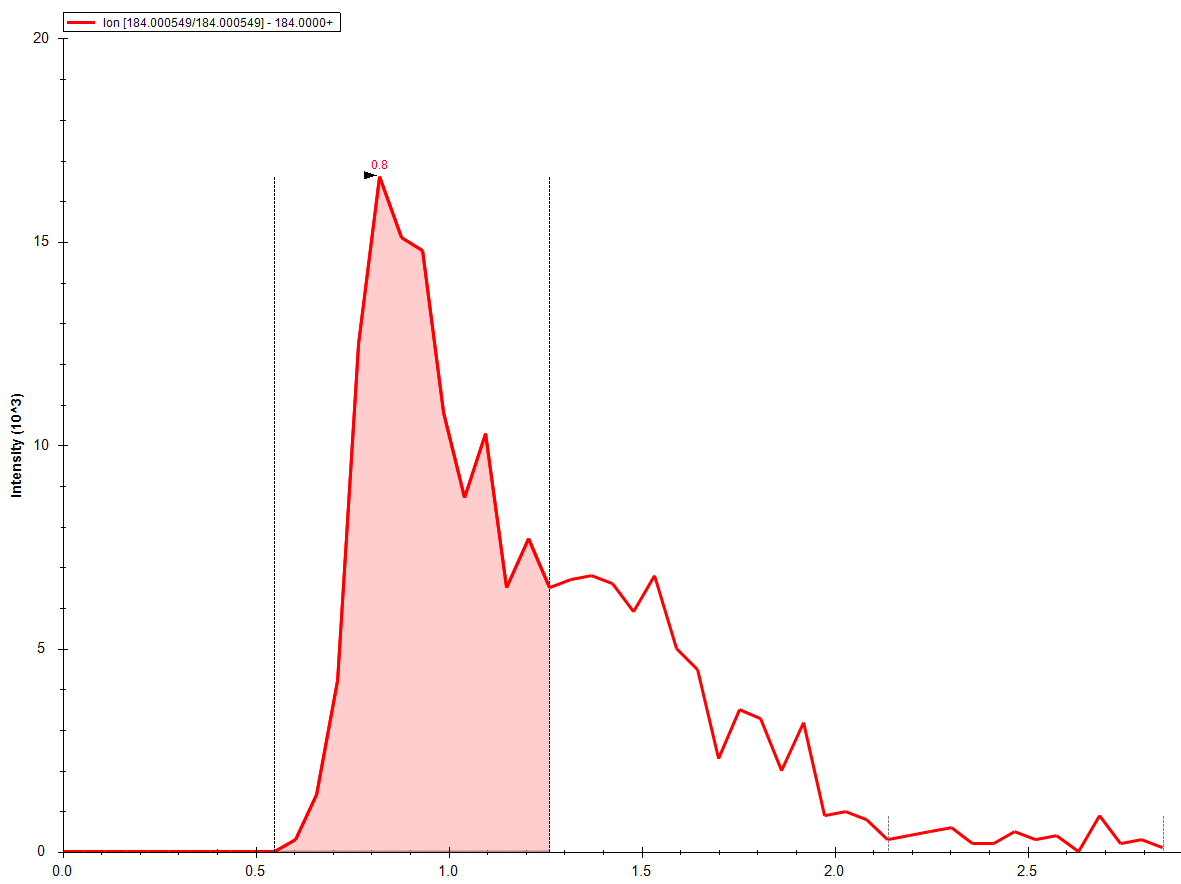 NeedsAdjustmentIntegration.PNG
NeedsAdjustmentIntegration.PNG error peak boundaries.png
error peak boundaries.png