Hi,
This is my first time using Skyline (v1.4), and I'm trying to use it for the MS1 filtering feature. I collected data on an orbitrap and have ended up with several files that I thought would work with Skyline to build a spectral library. First, I tried using a .pep.xml file exported from Proteome Discoverer. I ended up with an error that read:
ERROR: Could not find spectrum file " " in current directory ERROR: No spectra were found for the new library.
So I thought that maybe the error was due to the fact that I had searched the raw data with sequest and mascot (???? I don't think this reasoning is correct). Anyway, I went to the mascot server and converted it to a .dat file. I was more successful in doing it this way, however when I went to review the spectral library, the retention times are missing (it says RT:0) and the basename (I guess...) doesn't match the raw file name that I eventually want to import.
See the attached screenshot and the .dat file.
Let me know if you can help.
Thanks,
Laura |
| |
| Brendan MacLean responded: |
2013-01-12 12:39 |
Hi Laura,
I would love to get one of your .pep.xml files. That should work. So, I would be interested in debugging why it isn't.
As for the .dat files, I would recommend you use MSConvertGUI, which you get when you install ProteoWizard ( http://proteowizard.sourceforge.net/downloads.shtml) with the "TPP compatibility" checkbox checked, to create your MGF files for input into Mascot.
The retention times and source files must be specified correctly in the MGF files you use to search with Mascot, if you want to see them showing up in your .dat files, and subsequently in your spectral libraries.
Hope this helps. I will send you place to post your .pep.xml files.
Thanks for taking the time to post this description of your experience using Skyline.
--Brendan |
| |
| Brendan MacLean responded: |
2013-01-14 14:58 |
Hi Laura,
I get an error message about not being able to fine the spectrum mzXML file:
ERROR: Could not find spectrum file LEE_T0_IMAC_CID7_5July12_3.msf[.mzML|.mzXML] in current directory, ../,../../.
But this is expected, because you didn't actually send me the mzXML file. I am not seeing the error you mention:
ERROR: Could not find spectrum file " " in current directory
Did you omit the file name, or do you mean you see " " verbatim?
On your system, is the required mzXML file in either:
the current directory
the parent directory (..)
the grandparent directory (../..)
That is what is meant by the message I have pasted above: the library builder was not able to find the mzXML file in any of these three directories. If it is somewhere else, you will want to copy or move the mzXML file to one of these locations.
Note that the library builder should be smart enough to try removing the .msf included in the same. So, it will actually look for a file in these locations:
LEE_T0_IMAC_CID7_5July12_3.msf.mzXML
LEE_T0_IMAC_CID7_5July12_3.msf.mzML
../LEE_T0_IMAC_CID7_5July12_3.msf.mzXML
../LEE_T0_IMAC_CID7_5July12_3.msf.mzML
../../LEE_T0_IMAC_CID7_5July12_3.msf.mzXML
../../LEE_T0_IMAC_CID7_5July12_3.msf.mzML
LEE_T0_IMAC_CID7_5July12_3.mzXML
LEE_T0_IMAC_CID7_5July12_3.mzML
../LEE_T0_IMAC_CID7_5July12_3.mzXML
../LEE_T0_IMAC_CID7_5July12_3.mzML
../../LEE_T0_IMAC_CID7_5July12_3.mzXML
../../LEE_T0_IMAC_CID7_5July12_3.mzML
After that, it will give up and issue the error I pasted.
Does that help answer why you see that error? Are you able to solve it on your system now? Or is there something more complicated going on?
Thanks for sending your file.
--Brendan |
| |
| leedward responded: |
2013-01-15 13:26 |
Hey Brendan,
I did just omit the spectrum file name in my post. Sorry about that.
I think the real issue is that I don't fully understand the Skyline workflow. Where in the tutorial does it say that the mzXML file must be present? And as for generating the mzXML files, do you use proteowizard for that? Sorry for the confusion, I thought I only needed the pep.xml file. Again, I think this was just a misunderstanding. I hope it will work once I figure out how to generate an mzXML file! |
| |
| leedward responded: |
2013-01-15 13:39 |
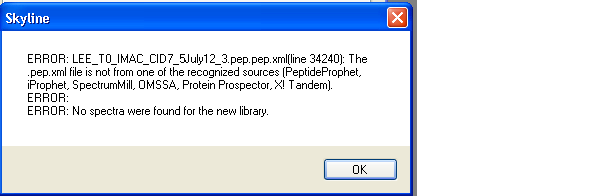
UPDATE:
I used proteowizard to generate the mzXML file and it is now in the correct directory. When trying to create a spectral library using the .pep.xml file, I now get a new error (see attached image). I will try to load the mzXML on the ftp server, but I think it might be too large (it timed out).
I went ahead and used proteowizard to generate a .mgf file that I attempted to submit to Mascot. However, Mascot gave me this error message:
"Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 6
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 22691
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 45376
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 58221
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 72086
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 82531
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 93156
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 106961
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 120586
Warning : Max number of ions is 10000. Ignoring ms-ms set starting at line 139311
Warning : Error 31 has been detected 4932 times and only the first 10 messages have been output"
So, it didn't really work? I then generated a .mgf file using proteome discoverer. This has been my most successful attempt thus far! After exporting the .dat output file, I used it to create a spectral library. Unlike previous attempts using .dat files (which were generated automatically with discoverer daemon 1.3), the retention times are now visible. HOWEVER, the name associated with the file is still "F010730.dat", instead of its base-name "LEE_T0_IMAC_CID7_5July12_3.raw". I am posting my .dat file to the ftp server right now.
Laura |
|
| |
| Brendan MacLean responded: |
2013-01-15 14:10 |
Hi Laura,
Sounds like PD produces MGF files with retention times but not source file information, and the daemon produces MGF files with neither. Unfortunate in both cases.
Returning to MSConvertGUI, I have attached a screen shot of the form, with an important setting highlighted. For the Peak Picking filter, you want to be sure to have Prefer Vendor checked. Otherwise, your MGF files may contain full profile MS/MS peaks, if your MS/MS were acquired in profile mode. Does that seem like it might explain why your MSConvertGUI generated MGF files might have more than 1000 peaks?
I am still looking ath the pepXML/mzXML case.
--Brendan |
|
| |
| Brendan MacLean responded: |
2013-01-16 17:31 |
Hi Laura,
I understand from your direct email to me that Mascot still complains about the number of peaks in the MGF files you are generating with MSConvertGUI, using the settings I posted. After further inspection, I have found that I missed a step: once you have the Peak Picking settings set up the way I showed, you actually need to click the Add button in MSConvertGUI to add the peak picking filter, before you do the conversion.
I have posted a new version of the PowerPoint slide showing how MSConvertGUI should be configured before you click the Start button.
Hope this will work for you. You will want the same settings for your mzXML file conversion.
I have a library builder that can import your pepXML and mzXML now, but in getting this to work, I was reminded that Proteome Discoverer writes out pepXML with broken modifications specified. See the end of this thread for details:
https://skyline.gs.washington.edu/labkey/announcements/home/support/thread.view?entityId=cc9bffa6-fbd8-102f-97b8-5d0a05bc6c8e&_anchor=2538#row:2538
So, you will want to continue working on the MSConvertGUI conversion of your RAW file to an MGF with proper RT and source file information. See the attached PowerPoint slide for details.
Sorry this has taken so long. Thanks for your patience.
--Brendan |
|
| |
| leedward responded: |
2013-01-17 08:04 |
Brendan,
Thanks for the information. I guess I didn't know how to generate the mgf/mzXML files correctly using Proteowizard. It seems that I won't be able to use PD generated pep.xml files to create spectral libraries, based on the other thread you sent me a link to. That is too bad considering that the other workflow (using mgf files) requires me to re-search my data with Mascot (not a huge deal, but still would've been nice not to have to re-search the data).
I went ahead and regenerated the mgf file then searched it in Mascot. The error that I was getting before does not show up anymore. I then attempted to create a spectral library with the .dat file and was successful! The base name and retention times are displayed now. Thanks again for being patient and helping me set up a spectral library.
Laura |
| |
| Brendan MacLean responded: |
2013-01-17 10:56 |
Hi Laura,
As is usually the case when the same issue crops up over and over, we are starting to think of work-arounds that would allow us to create valid spectral libraries from the Proteome Discoverer pepXML, which does not follow the pepXML specification for its amino acid modifications. So, we may have a fix for that in the near future.
We are also going to increase the priority on our effort to make it possible to build spectral libraries directly from .msf files. Hopefully, we will be able to release that feature in the next month or two.
Thanks for your feedback, and your willingness to try a work-around until we can make support for your preferred workflow more seamless.
--Brendan |
| |
|
|
 LEE_SpectralLib_error_15Jan13.bmp
LEE_SpectralLib_error_15Jan13.bmp