| |
| Brendan MacLean responded: |
2015-11-25 14:34 |
Hi Brett,
At present, this is not available. I have posted a TODO issue to the Skyline issues list to review the Edit > Refine > Advanced form and add new filters to it, including this one. Hopefully we can get to this in the first 2016 release cycle.
At the moment, you are stuck with manual use of right-click > Remove Peak in the chromatogram graphs, or if you create a mProphet peak scoring model, you can choose to remove all peaks above a q value cut-off during Edit > Refine Reintegrate.
Thanks for your feedback.
--Brendan |
| |
| Brendan MacLean responded: |
2015-11-25 14:34 |
|
| |
| robwsprung responded: |
2017-09-15 07:06 |
Hi Brendan,
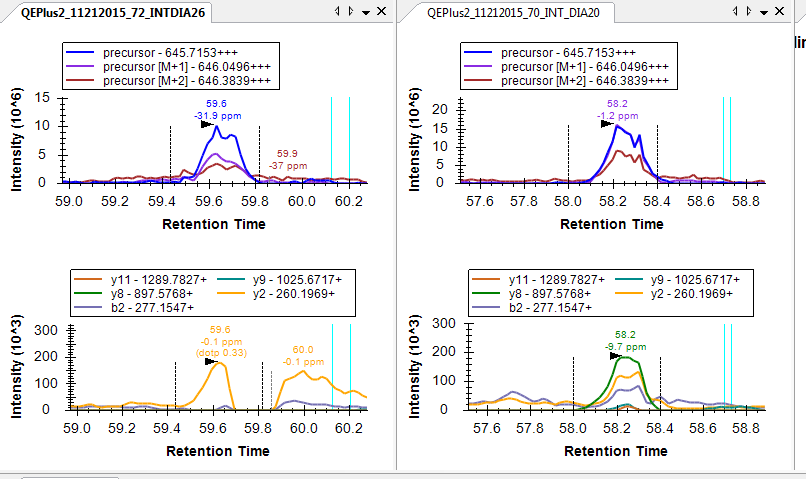
I am working with PRM data and would find such a product-ion mass error filter useful as well. We are performing isotope dilution experiments in a variety of matrices, including patient samples, using Q-Exactive instruments.
I observe variable levels of interference in different channels among matrices, often associated with high mass error (>20ppm). Having a mass error filter would be helpful to exclude the interference and facilitate selection of a robust set of transitions to use for quantitation across a patient cohort.
Thanks for your continued development of this excellent software.
Robert |
| |
| Brendan MacLean responded: |
2017-09-23 17:28 |
Hi Robert,
I would now recommend giving the "Centroided" option a try, instead of using "Orbitrap" on profile-mode spectra. When you switch to "Centroided", you will be able to set a mass accuracy tolerance, with which I have had good luck using between 10 and 20 ppm.
This might work very well for your application. It is working so well for us that some of the researchers in the MacCoss lab are no longer keeping profile mode spectra in their raw data files, and instead storing centroid-only raw files, which saves on disk space.
Let me know what you think if you give it a try. Sorry for the wait on a response. Busy at HUPO last week.
--Brendan |
| |
|
|
 skyline1.png
skyline1.png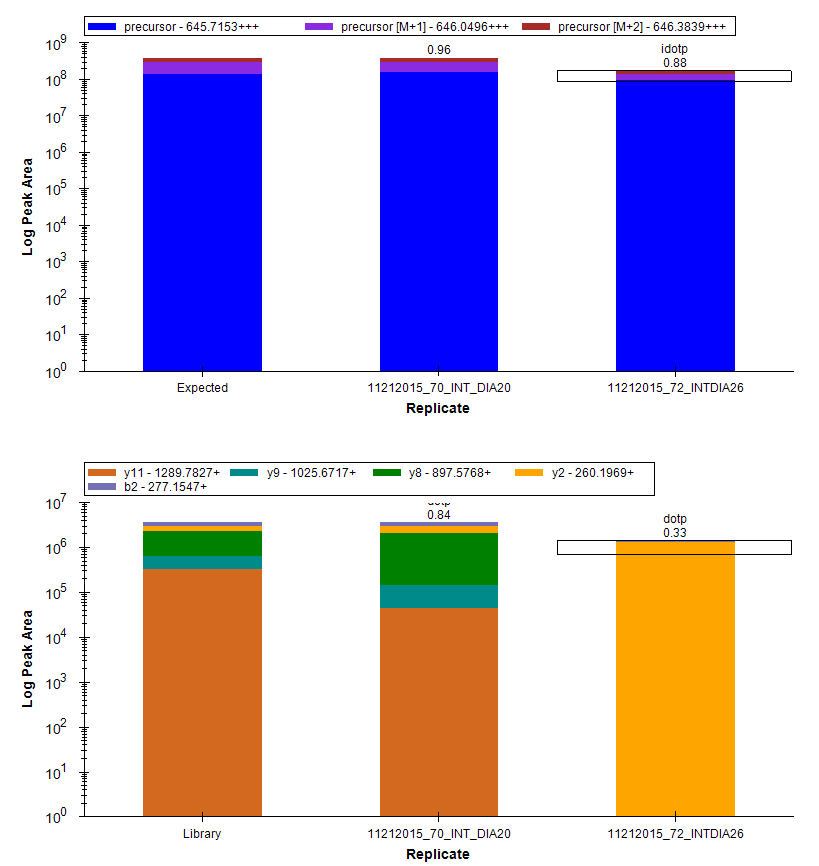 skyline2.png
skyline2.png