Hi Brendan,
Thanks for the 3.1 release! I was wondering whether there is a way to normalize label-free data against the TICs of DIA MSMS runs (I get those using Bruker DataAnalysis). I have entered a separate column in the Skyline replicates table with the relative TIC integrals for each replicate. But I cannot select that column for normalization under group comparisons. It only shows up as an option for control group annotation and identity. It would be nice to have the option to select this column of DIA TIC values for normalization. I have been using the DIA TICs to normalize each transition in a sample/ replicate-specific fashion to account for small differences in loading or sample ionization after exporting the data to Excel. But with the new group comparison that includes statistics it would be great to be able to do it within skyline to save time.
Thanks for any suggestions,
Dietmar |
| |
| Brendan MacLean responded: |
2015-03-16 16:18 |
Hi Dietmar,
Interesting idea. Skyline does now store the TIC chromatogram for full-scan experiments. I am not that familiar with this mode of normalization. Are you saying you just sum up all intensity measures in the TIC, which seems like it would be dependent on sampling frequency, or are you taking an area under the curve of the entire gradient? Or something else?
I would like to understand better. It seems like we might be able to help, either by offering this as a choice, or, as you suggest, by allowing you to normalize by a replicate annotation value, which also seems reasonable.
Thanks for your feedback.
--Brendan |
| |
| tobias.kockmann responded: |
2019-09-04 01:35 |
Hi Brendan, Hi Dietmar,
I also have a case here where a TIC-based standardisation might be beneficial. The data was acquired using PRM incl. heavy std. peptides. By looking at the TIC I actually realised that the total sample amount was not properly adjusted before digestion (TIC area shows a high fluctuation from sample to sample, std. peptide signals are very stable).
Does anyone have a reference how TIC-based standardisation should be performed in this case? I was thinking to export to MSstats and do this manually prior to dataProcess(...) by scaling each light signal to def. unit of TIC area. As Brendan pointed out the TIC total area is reported by skyline. Does this have any downstream affects on statistical testing?
Greetings,
Tobi |
| |
| Nick Shulman responded: |
2019-09-05 11:18 |
Tobi,
If you want to do TIC normalization in Skyline, you can take advantage of the feature where Skyline lets you specify the "Explicit Global Standard Area" to use for each replicate.
Here's the instructions from https://skyline.ms/announcements/home/support/thread.view?rowId=35816:
When Skyline extracts chromatograms from MS1 data, Skyline remembers the total ion current.
You can see this value in the Document Grid.
It's under:
Replicates > Files > Total Ion Current Area
This column will contain the integral of the Total Ion Current of all of the MS1 scans.
Right next to the Total Ion Current Area column is the "Explicit Global Standard Area" column.
If you want to normalize to an arbitrary number, then you can type that number into the Explicit Global Standard Area column. Then, whenever you ask Skyline to normalize to Global Standards, Skyline will use that number instead.
A common thing to do is to copy the values from the Total Ion Current Area column and paste them into the Explicit Global Standard Area column.
(It has been suggested that we should add "TIC" as one of the normalization methods along with "Global Standards", "Equalize Medians" etc.)
|
| |
| wphipps5 responded: |
2024-08-21 13:58 |
Sorry this is an old post but I have a related question-- "This column will contain the integral of the Total Ion Current of all of the MS1 scans." << Does this mean TIC including only those precursors extracted in Skyline (precursors on your target list, excluding iRT standard peptides)? I noticed for some DIA files, the Total Ion Current reported in Skyline is less than the sum of the Total Areas for spiked iRT peptides, which is only possible if iRT peptide areas are excluded from determination of TIC
Thanks,
Bill
|
| |
| Nick Shulman responded: |
2024-08-21 14:33 |
There are two different ways that Skyline might calculate the Total Ion Current Area value that you see in the Document Grid.
If the mass spec data file contains a TIC chromatogram, then Skyline will calculate the Total Ion Current Area by taking the integral under that chromatogram.
The TIC chromatogram usually contains TIC values for all types of spectra, but Skyline only looks at the points which came from MS1 spectra.
If the data file does not contain a TIC chromatogram, or if Skyline is not able to figure out which points in the TIC chromatogram came from MS1 or MS2 then Skyline falls back to the slower way of calculating the Total Ion Current Area which involves looking at every MS1 spectrum and adding up all of the intensities in that spectrum.
If Skyline calculates the Total Ion Current Area in the slow way by looking at each spectrum, then I would expect that the Total Ion Current Area would be much larger than the areas of your spiked in iRT peptides.
If Skyline is calculating the Total Ion Current Area using the TIC chromatogram then it is difficult to predict the relationship between the Total Ion Current Area and the areas of your peptides. The intensity values in the TIC chromatogram are whatever the mass spectrometer said that they were. The total ion current values reported by the mass spectrometer are expected to be proportional to what Skyline thinks they are, but they could be orders of magnitude different because there are multiple ways to sum up all of the intensities. (I am not sure, but it might have something to do with whether you multiply or not by the delta m/z when summing the rectangles).
If you want to force Skyline to calculate the Total Ion Current Area in the slow way, you could convert your raw file to .mzML and tell msconvert.exe to discard all of the chromatograms.
The way to tell msconvert.exe to discard all of the chromatograms is to use the "chromatogramFilter" and specify a chromatogram index which does not exist.
msconvert.exe --chromatogramFilter "index 999" myrawfile.raw
You can certainly send us your data if you would like more information about how Skyline is coming up with the numbers that it is reporting.
In Skyline you can use the menu item:
File > Share
to create a .zip file containing your Skyline document.
The Share Document dialog gives you the option to include raw files, or you could zip them up and send them to us separately.
Files which are less than 50MB can be attached to these support requests. You can always upload larger files here:
https://skyline.ms/files.url
-- Nick
|
| |
| wphipps5 responded: |
2024-08-22 14:10 |
Thank you, Nick. I uploaded an example Skyline document (wphipps5_20240822_DIA-ATTR.sky.zip) and file (0021-240213-A1-THY.mzML).
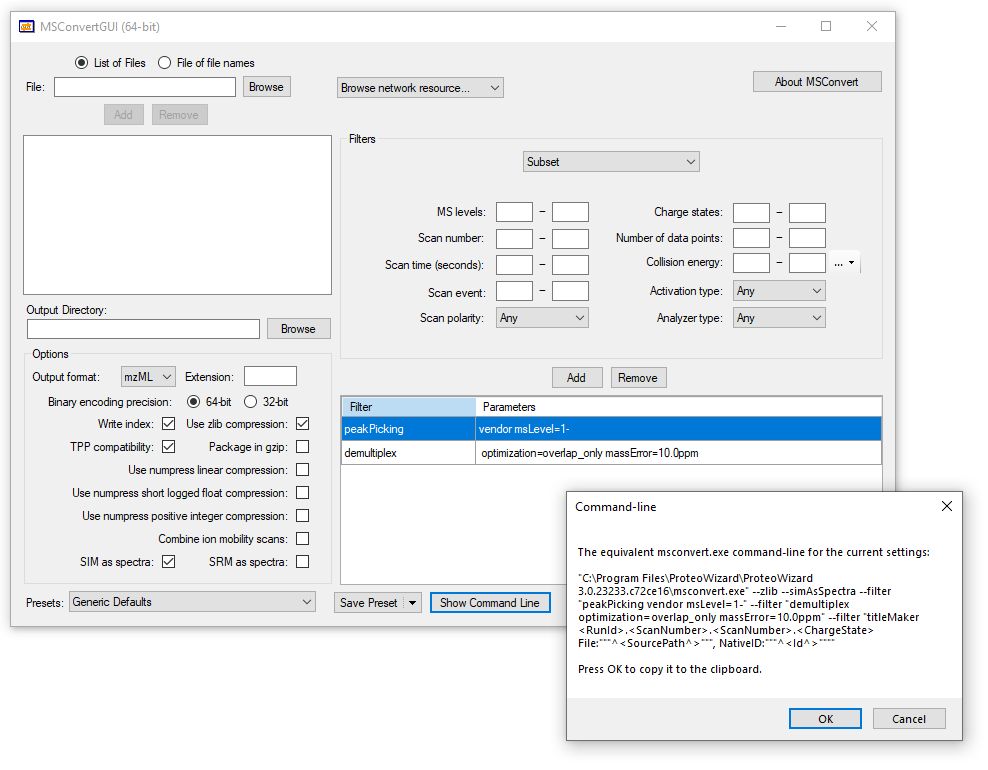
Our DIA acquisition employs 24 m/z wide overlapping windows. Normally I de-multiplex these files and convert them to mzML in the GUI version of MSConvert (screenshot attached) before importing to Skyline. I am not great at using MSConvert at the command line but the GUI version does not include the chromatogramFilter so I would need to use the command you've written. Can I simply append the chromatogramFilter after peakPicking and demultipex filter commands?
ChatGPT recommended me the following:
"C:\Program Files\ProteoWizard\ProteoWizard 3.0.23233.c72ce16\msconvert.exe" --filelist filelist.txt --zlib --simAsSpectra --filter "peakPicking vendor msLevel=1-" --filter "demultiplex optimization=overlap_only massError=10.0ppm" --filter "titleMaker <RunId>.<ScanNumber>.<ScanNumber>.<ChargeState> File:"""^<SourcePath^>""", NativeID:"""^<Id^>""" --chromatogramFilter "999" -o D:\DIA-Amyloid\output
Thanks,
Bill
|
|
| |
| Nick Shulman responded: |
2024-08-22 15:11 |
Bill,
Thank you for uploading those files. I will take a look at them soon.
It might also be helpful if you could upload the .raw file "STMS_20240221_23.raw" that the .mzML file came from.
Yes, you can add the chromatogram filter onto the end there, but it looks like ChatGPT is missing the word index that really does need to be there.
If you already have a .mzML file that you like you can use msconvert to remove all of the chromatograms by doing something like:
msconvert.exe 0021-240213-A1-THY.mzML --outfile myNewFile.mzML --chromatogramFilter "index 999"
-- Nick
|
| |
| wphipps5 responded: |
2024-08-22 16:04 |
Thank you. I uploaded the corresponding raw file. I tried applying the chromatogramFilter using the command you provided, and imported the result into the same Skyline document. I put the two files next to each other in the document (before and after applying chromatogramFilter, screenshot attached). To my surprise the total ion current area did not change (I was hoping it would get bigger). Does this mean Skyline was already calculating total ion current area the "slow" way?
For context, the whole reason for doing this is that I am seeking some way to subtract iRT peptide area contribution out of the TIC value. We're normalizing MS2 total area for certain peptides to the TIC across samples but iRT peptides were not added evenly to all samples.
Thanks,
Bill
|
|
| |
| Nick Shulman responded: |
2024-08-22 17:12 |
Oops.
I had forgotten that the actual reason that the Total Ion Current Area ends up being much lower than you would expect it to be compared to the peak areas of all of your peptides is that the peptide peak areas have all been multiplied by 60.
Basically, the Total Ion Current Area is in units of current times minutes, whereas all of the other peak areas you might see in the Document Grid are in units of current times seconds.
If you multiply the Total Ion Current Area values reported by Skyline by 60 then you do end up with numbers which are probably in the expected range compared to the MS1 areas of your iRT peptides.
I think a more conventional normalization approach if your iRT peptides cannot be used for normalization, would be to normalize against some endogenous peptides from proteins that biologically do not usually change between samples.
-- Nick |
|
| |
|
|
 screenshot.PNG
screenshot.PNG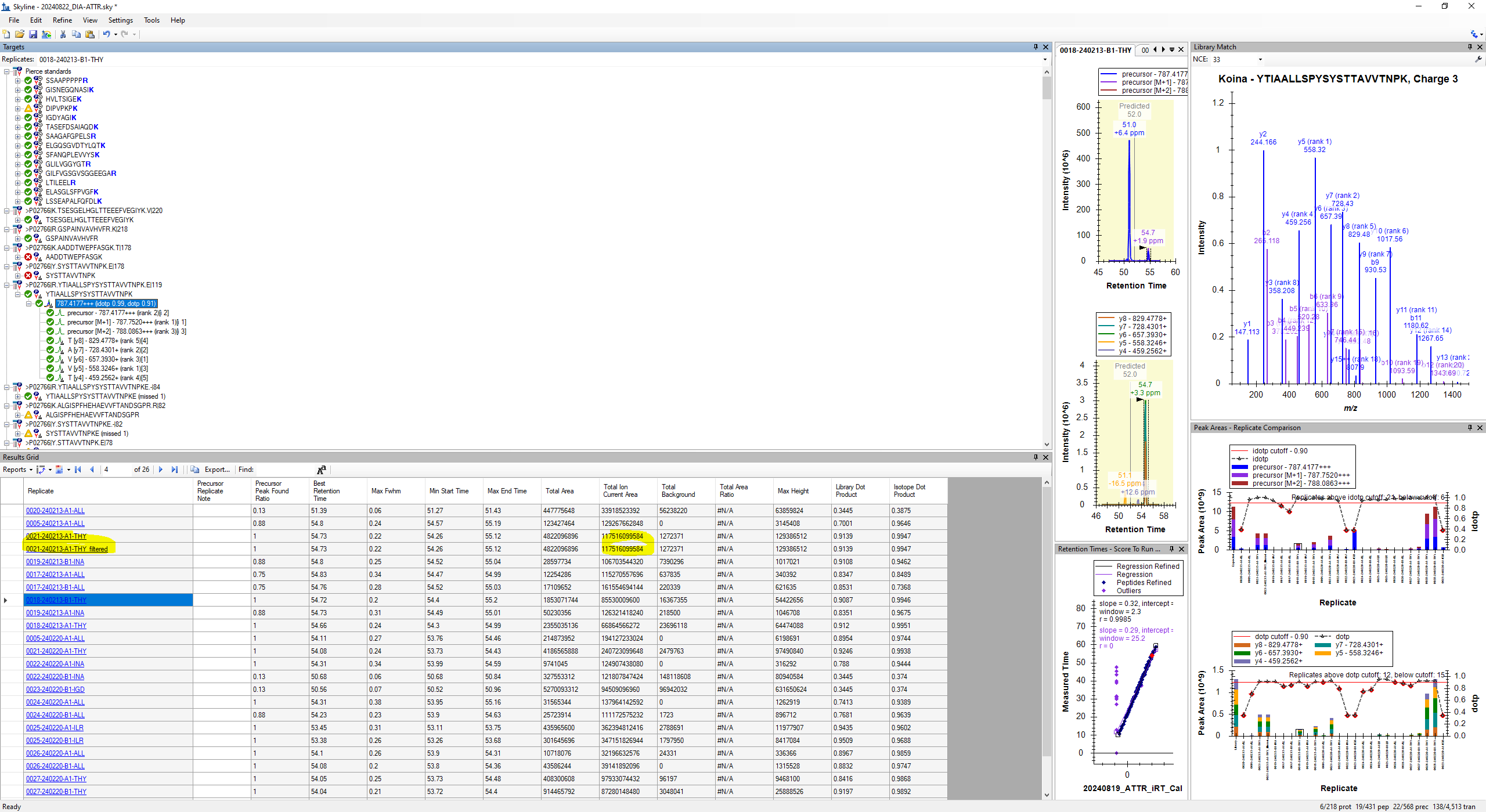 screenshot.PNG
screenshot.PNG