We are working with DIA acquisitions for targeted protein quantification and try to figure out the easiest way to assemble a complete and best quality spectral library based on different DDA discovery analyses.
The BlibFilter algorithm chooses the best spectrum based on the best score. However, we have spectral libraries produced from different search engines, e.g. a pride.xml export from PeptideShaker with several search engines (X!Tandem, Amanda, msgf+, etc.), MaxQuant, and ProteomeDiscoverer.
How is the best score criteria applied in this case? By trying to build a spectral library using a pride.xml file containing X!Tandem and Amanda data together with MaxQuant, we realized that most of the library came from this pride file but the quality of the fragment spectra was miserable. I also realized that in this case, the "Filtering redundant library" process during the "Building Peptide Search Library" task was not performed, while it was done when using only data coming from one search engine (MaxQuant).
Hence, I guess it is not possible to build a spectral library using data from different search engines, is it?
Alternatively, the BlibFilter chooses the spectrum with the highest TIC. Based on above observation (and others), I am not convinced that this works really well.
Would it be possible instead of choosing the best spectrum, to compile all MS2 spectra belonging to the same precursor (charge and m/z values) to one single spectrum by adding all available intensities? Unfortunately, this might have a drawback, because not all fragment mass values will have the exact same digits after the coma?
I am looking forward to your answer or suggestion for dealing with this issue.
Thanks a lot and best wishes,
Manfred |
| |
| Kaipo Tamura responded: |
2014-11-07 10:31 |
Hi Manfred,
It is correct that BlibFilter breaks score ties using TIC. In the case of multiple score types, BlibFilter will gather the spectra with the highest score for each score type, then select the spectrum with the highest TIC among those winners. We have also experimented with choosing spectra using dot products (this is actually the default when running BlibFilter from the command line when the 'best-scoring' flag is not set; Skyline sets this flag) and consensus spectra, but there were issues with both of these approaches.
I wouldn't mind adding this summed spectra approach as another option (i.e. it would be used then BlibFilter is called using a command line flag). We could solve the issue of different precisions by rounding all of the values to the least precise one, but there might be a few other details to work out. For example, would it be a good idea to normalize the spectra somehow and/or weight them differently when creating a summed spectrum.
Thanks,
Kaipo |
| |
| manfred.heller responded: |
2014-11-10 07:14 |
Hi Kaipo
Thanks for the clarifications. Unfortunately, it does not explain the redundant library filtering step, that does seem to be done only, when several peptide search results from the same search engine are loaded. It's not happening when I load results from different search engines.
The TIC choice does somehow make sense. However, we had stumbled by accident over a case where the more intense spectrum, which was chosen for the library, was of worse (fragment) quality than a less intense one. This particular peptide produced a high intense y2 ion and in the more intense spectrum some higher y-ions were missing while present in the less intense spectrum. This observation made me thinking of the summing approach.
Why do you think that a normalization or weighting of the different spectra is necessary? The above described case shows somehow, that there is no golden standard fragment spectrum. Summing, or averaging, intensity values might be a way of approaching the representation of the golden standard spectrum. Of course, I'm talking here only about data coming from the same type of MS analyzer.
Best wishes,
Manfred |
| |
| Kaipo Tamura responded: |
2014-11-10 10:04 |
Hi Manfred,
The filtering step should not be affected by results from different search engines, so that may be a bug. Would you mind sharing an example of this data via email?
Regarding the weighting, I was thinking that it may produce better results if we give less consideration to low quality spectra (e.g. low signal-to-noise ratio). However I am certainly not an expert on this subject and introducing complexity can always cause other problems.
Thanks,
Kaipo |
| |
| manfred.heller responded: |
2014-11-11 05:15 |
Hi Kaipo,
I made screen shots from the skyline user interface.
The Bad_SpecLib_CD14 is the example where skyline preferred to use this spectrum over Good_not_selected_SpecLib_CD14 (as explained in my previous mail). The Bad came from a MaxQuant (Andromeda) identification while the Good was from a ProteomeDiscoverer (Sequest) search result. Please note, that the data came from different samples with approximately four times more material loaded for the LC-MS/MS run in case of the Bad spectrum. In addition, the Bad spectrum was recorded with a relative collision energy of 30 while the Good was with 27 (both on the same QExactive instrument).
Regarding my comment about the golden standard of a fragment spectrum, I have also attached two spectra from the same peptide (same data set as above) with different relative fragment ion intensity distributions resulting in different ranking of peaks (files diff_Quality_library...).
Since we started this discussion, we tried to build a master library from about 140 LC-MS/MS runs. I have attached a striking example (one of many) where skyline has chosen the worst spectrum for 3+ peptide SWAELQQWLKPGLK (File Library_winner). I included 4 out of many other spectra that were in this "competition" and are clearly of superior quality but not chosen as winner (Library_competitor1 etc.). Furthermore, Skyline has repeated the same strange choice on the 2+ ion, as mentioned above (Bad_SpecLib...). Hence, I'm not convinced that the BlibFilter is working well.
One note to the weighting issue: Seeing these examples convinces me even more that the averaging/summing idea should be implemented. One could normalize each fragment spectrum by its base peak before summing up peak intensities.
I hope this helps,
Manfred |
|
| |
| kreimer s responded: |
2018-09-25 04:33 |
I have a related issue, also trying to build a spectral library from a PD2.2 .pdresult file in the most recent Skyline-daily release. The library is built but for some reason many poor quality spectra are selected instead of the high quality spectra which were used for peptide identifications. By poor quality, I mean only one fragment ion matched out of many. Not sure if this is relevant but the data is from a SILAC experiment. I've tried this while setting the cut-off value to 0.95 and 0 with the same result. This is from a 48 run dataset. |
| |
| Brendan MacLean responded: |
2018-10-17 13:55 |
Just noticed this cleaning out my inbox. Can you try again with the latest Skyline-daily, and if it is still an issue, please post to a new thread. This one is 4 years old. |
| |
|
|
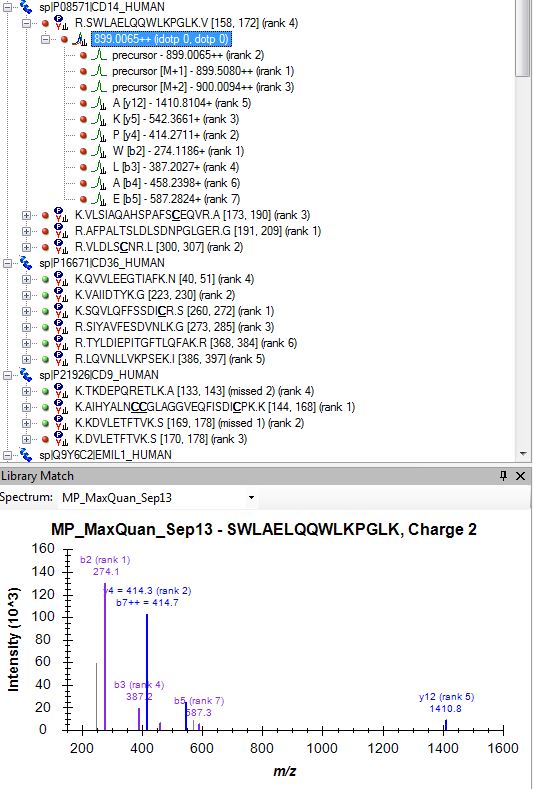 Bad_SpecLib_CD14.JPG
Bad_SpecLib_CD14.JPG Good_not_selected_SpecLib_CD14.JPG
Good_not_selected_SpecLib_CD14.JPG diff_Quality_library_CD14_PSM1.jpg
diff_Quality_library_CD14_PSM1.jpg diff_Quality_library_CD14_PSM2.jpg
diff_Quality_library_CD14_PSM2.jpg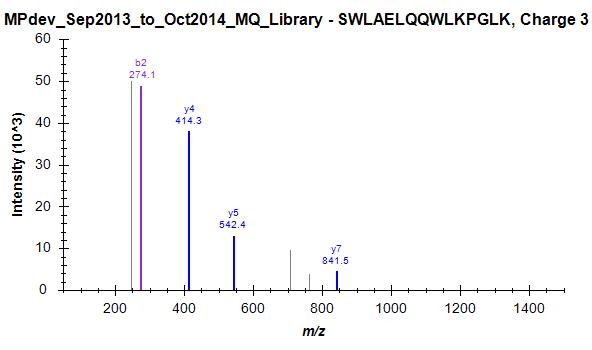 Library_winner.jpg
Library_winner.jpg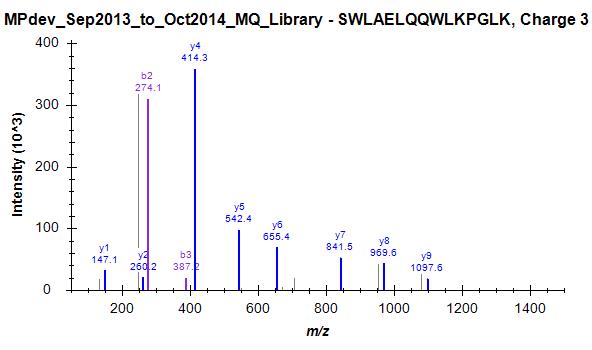 Library_competitor1.jpg
Library_competitor1.jpg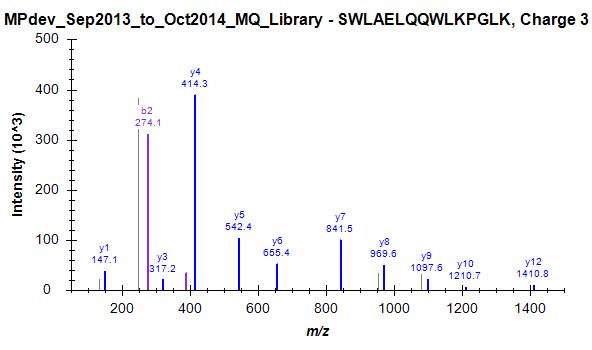 Library_competitor2.jpg
Library_competitor2.jpg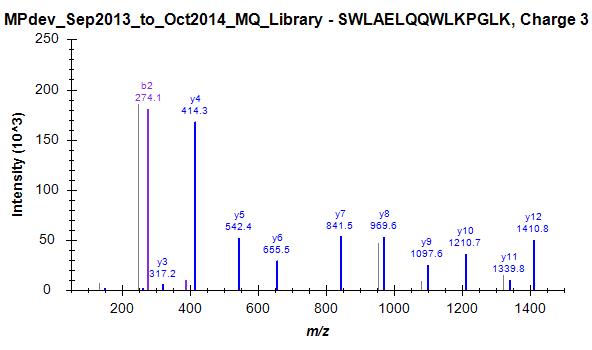 Library_competitor3.jpg
Library_competitor3.jpg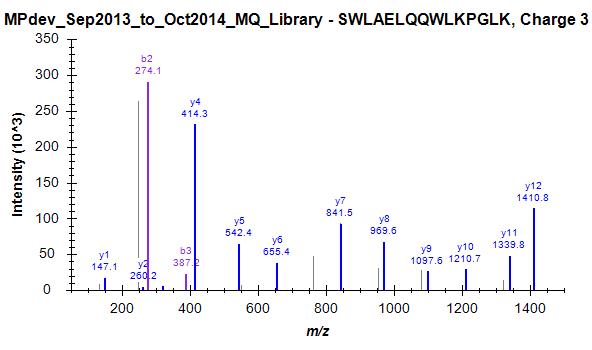 Library_competitor4.jpg
Library_competitor4.jpg