| |
| Brendan MacLean responded: |
2012-10-19 15:26 |
Hi Lei,
This indicates that you have not given Skyline enough information to be able to understand the modification state of your peptides. It would be useful to see the contents of your Peptide Settings - Modifications tab.
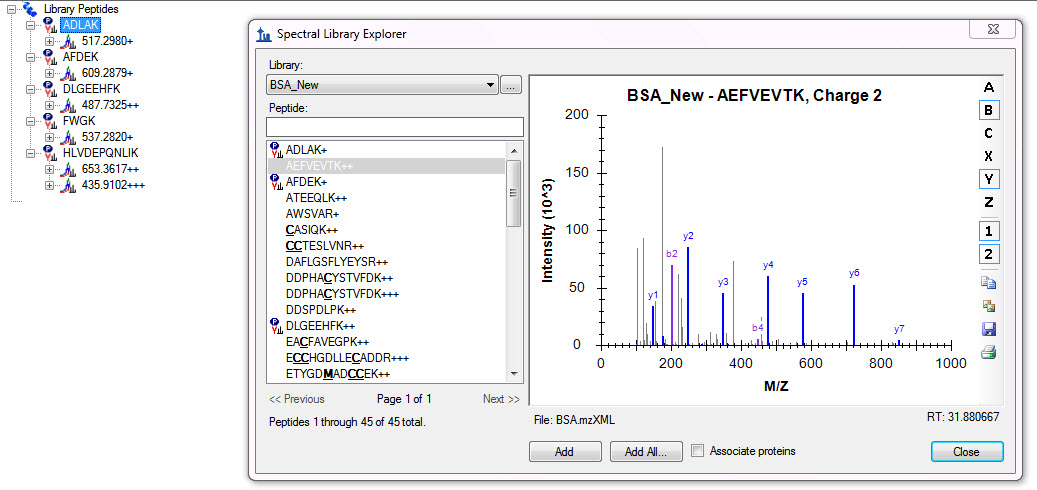
From the image you sent, it is easy to guess why Skyline might not be able to figure out the modifications on peptides with modified Cysteines and Methionines. A little harder to guess why it would have a problem with peptides that appear unmodified, unless you have defined static modifications that it expects on those peptides.
Usually, when you first bring up the Spectral Library Explorer it will attempt to figure out modifications it might apply to make sense of the modified peptide sequences in your library, and it will ask you whether you want to accept to modifications it guesses. I would expect it to be able to guess oxidized Mehtionine and carbamidomethyl Cysteine, but you have have some other Cysteine modification or this may be some other Cysteine modification.
The easiest way to fix this is for you to use the Peptides Settings - Modifications tab to manually define the modifications you used in your peptide search.
If you post your modification settings, I may be able to explain better. Also if you hover the mouse cursor over a peptide in the Spectral Library Explorer with a bold, underlined amino acid character, Skyline will display a tip with the modification mass encoded within the sequence, which can be helpful in verifying that modification masses are what you expect. I have seen searches made with average masses fail to match with Skyline settings that indicate monoisotopic masses should be used.
Hope that helps.
--Brendan |
| |
| lei_wang responded: |
2012-10-19 18:36 |
Hi Brendan,
Thank you very much for these head-ups. I will send you the image of my modifications tab when back in the office. Indeed, alkylation of cysteine in this sample was done by iodoacetic acid which produce a 58, but not 57Da mass shift. I will add in this modification and try it again.
Another question: is Skyline, or will Skyline be, supporting high resolution SIM scan or metabolomics LC-MS data analysis which no database search is involved?
Thanks again and have a nice weekend,
Lei |
| |
| lei_wang responded: |
2012-10-22 07:54 |
Hi Brendan,
I changed cycteine alkyation in peptide setting but still could not add those peptides from the spectral library. I have included all the screenshots in the attached ppt file. So in peptide setting modification tab, how to define a modification is static (cycteine alkylation) or dynamic (methionine oxidation, STY phosphorylation etc)? Should I just leave the dynamic unchecked?
Thanks,
Lei |
|
| |
| Brendan MacLean responded: |
2012-10-22 08:05 |
Hi Lei,
Your most obvious immediate issue is that you need to uncheck the "Carbamidomethyl Cysteine" modification, since having it checked is causing Skyline to expect both that and "Carboxymethyl (C)" on your C residues.
I am not quite sure what is going on with your Methionine modification.
But, really, Skyline should provide you with all the information you need to decipher these kinds of issues for yourself. If you hover over the peptides that Skyline is not recognizing, you will see the modification masses encode in the sequence, which Skyline is failing to interpret. Then you just need to take a close look at your modification settings (and possibly your mass type - monoisotopic v. average) in the Transition Settings - Prediction tab. And figure out why those settings are failing to produce the delta masses you see in the Spectral Library Explorer.
At least if you uncheck "Carbamidomethyl Cysteine" Skyline should start recognizing you Cysteine modifications.
Good luck. Thanks for posting all the screenshots. If you still can't figure out your M modification, you will need to post a screenshot of the tip you get when you hover the mouse cursor over the peptide with the modified M.
--Brendan |
| |
| lei_wang responded: |
2012-10-22 08:19 |
Hi Brendan,
Thank you very much for your quick reply!
I hover over one of the peptide which has both M and C. It is very strange which show M modification is -115 and C is -45. I did remove the carbamidomethyl cysteine and only kept Carboxymethyl (C), Oxidation (M), Phospho (Y) and Phospho (ST).
Lei |
|
| |
| Brendan MacLean responded: |
2012-10-22 08:28 |
Hi Lei,
Those really are the modifications the library builder is attributing to those peptide matches. So, that leaves three possibilities:
1. The library builder is misinterpreting the modifications in your peptide search results.
2. The .msf to pepXML converter is not interpreting the modifications correctly, and is writing out bad pepXML.
3. Proteome discoverer somehow is actually set up to allow those modifications, and PD believes it is finding them.
So, probably time to dig into your search results a bit more. The pepXML should be pretty easy to open up in a text editor and look at the modifications on these peptides. Are they something other than what is being shown in the Library Explorer? If so, then I will need to get the pepXML from you to understand whey the library builder is not interpreting them correctly, but more likely it will turn out to be something in either PD itself or the PD to pepXML converter.
Good luck. Interested to hear what you find.
--Brendan |
| |
| lei_wang responded: |
2012-10-22 09:28 |
Thanks Brendan,
Here is the pepXML file exported from PD (I used Sequest). Although PD has another workflow node "spectrum exporter" to export the msf file to an mzData or mzML file, I have not tried those.
Best,
Lei |
|
| |
| Brendan MacLean responded: |
2012-10-22 09:51 |
Hi Lei,
Yes, the problem is in your pepXML. Your modifications show up as:
<mod_aminoacid_mass position="2" mass="58.005479" />
This is a misinterpretation of what the "mass" attribute for the <mod_aminoacid_mass> tag is supposed to mean. The mass tag is supposed to contain the total mass of the modified amino acid residue, not the modification delta-mass.
This might be a little clearer, if the pepXML actually contained global modification definition tags in the <search_summary> tag. Here is an example of what that might look like:
<aminoacid_modification aminoacid="C" massdiff="57.100000" mass="160.109185" variable="N"/>
This tag contains both the "mass" and "massdiff" attributes. The <mod_aminoacid_mass> tags, however, should contain only the "mass" attributes, and they should match.
So, yes, I think your problem stems from a bug in the converter you are using to produce your pepXML.
You'll have to work with Thermo on getting that fixed, or find another way to export your pepXML. Alternatively, if you use Scaffold, you can export mzIdentML and MGF files from Scaffold, which others have had success with.
Good luck. Thanks for your time in digging into this and supplying such useful information.
--Brendan |
| |
| lei_wang responded: |
2012-10-22 10:35 |
Hi Brendan,
Thank you very much for this detailed explanation. I now understand that there is a bug in the converter inside PD. Except for Scaffold, do you know whether there are some other small standalone programs which can convert thermo msf file to the format Skyline can recognize?
Thanks again,
Lei |
| |
| Brendan MacLean responded: |
2012-10-22 15:14 |
Hi Lei,
Unfortunately, I do not have another alternative. Others have reported success in using pepXML/mzXML from Proteome Discoverer, but I can't see how they could have been interested in PTMs, given what you have shown about the way PD exported pepXML files report modifications.
We do hope to support .msf files directly eventually. It would be great if you could report this to Thermo, though. I would certainly be interested in the response you get, whether they can fix it, and if so, how quickly.
Sorry I can't be more help in this case.
--Brendan |
| |
| lei_wang responded: |
2012-10-22 19:24 |
Thanks Brendan,
I will report this to Thermo. I will forward their reply to you if I get any. Really appreciate it very much for your help in this case!
Lei |
| |
| lei_wang responded: |
2012-10-24 14:48 |
Hi Brendan,
I sent the question to Thermo yesterday but so far there is no reply. I guess there will be no help from them.
I used another program "thermo-msf-parser" which can convert msf file to a csv file or mgf file. I checked the csv (attached) and PTMs are correct. Do you think Skyline can use either of these two files to build up the spectral library?
Thanks,
Lei |
|
| |
| Brendan MacLean responded: |
2012-10-24 15:03 |
Hi Lei,
I think you are best off waiting on this one until you either get a fix from Thermo, or we implement direct support for MSF files. You can track this item in the issues list:
https://skyline.gs.washington.edu/labkey/issues/home/issues/details.view?issueId=190
Your pointer to the MSF project should be helpful in getting feature implemented. It is much more interesting to us than adding support for arbitrary CSV formats. Unfortunately, it is not yet at the top of our priority list, but hopefully it won't be that long until someone here gets to it.
Thanks for digging into this. Sorry I can't offer more help sooner.
--Brendan |
| |
| lei_wang responded: |
2012-10-24 17:33 |
Sure thanks Brendan,
I will wait. Not in a hurry. Please take your time.
All the best,
Lei |
| |
|
|
 spectral library.jpg
spectral library.jpg M_C_modification.jpg
M_C_modification.jpg