| Brian Pratt responded: |
2023-02-22 12:57 |
Thanks for providing all the files.
That's unusual data - for MRM we'd expect to see chromatogram data rather than full scan. And for full scan data we'd expect to see MS1 data along with the MS2 data.
Is there a different way to export the data?
Thanks for using the Skyline support board,
Brian Pratt
|
| |
| ho-tak lau responded: |
2023-03-08 13:09 |
Hi Brian,
Sorry for the late reply. I have been struggling with changing the method. I also tried to build a prmPASEF method by adjusting the default Bruker prm method just to fit the molecules I am interested in, but the data look the same as the MRM data.
What do you mean by exporting the data in a different way?
Ho-Tak
|
|
| |
| Brian Pratt responded: |
2023-03-08 13:41 |
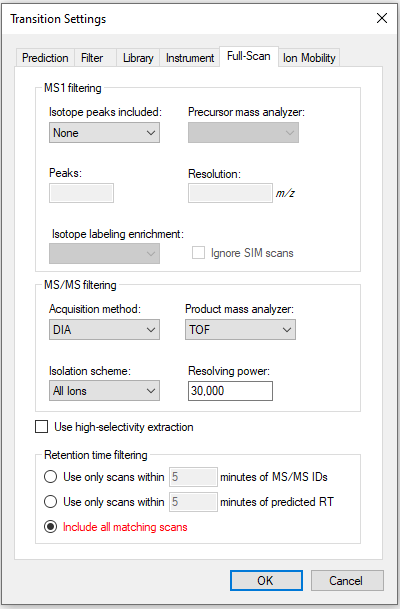
I think I just didn't look at your settings closely enough. Have a look at the attached image and see if that works better for you.
Best regards,
Brian
|
|
| |
| ho-tak lau responded: |
2023-03-08 14:21 |
Thank you! It works now.
I also want to report a parsing bug without opening a new thread.
When I tried to build library from Fragpipe 19.1 interact-xxx.pep.xml with the xxx_uncalibrated.mgf using Skyline 22.2.0.351. Skyline reported an error, I have to rename the .mgf to xxx_uncalibrated_uncalibrated.mgf.
Ho-Tak
|
| |
| Brian Pratt responded: |
2023-03-08 14:28 |
It's really better to open a new thread, if you'd be so kind.
Thanks!
Brian
|
| |
| ho-tak lau responded: |
2023-03-09 14:53 |
Hello Brian,
What is the difference between PRM and DIA in Transition Settings -> MS/MS filtering. I have another set of data with two molecules. Using PRM, Skyline will only pick up 1 molecule, but when I switch to DIA, All ions. It will pick up both molecules.
|
|
| |
| Nick Shulman responded: |
2023-03-09 15:32 |
Ho-Tak,
I see that in your Skyline document that you have a precursor with the m/z "873.2409".
I also see spectra in your Bruker .d folder where the isolated precursor was "873.3297".
When you choose "PRM" as the acquisition method, Skyline will extract a chromatogram point from a spectrum if the m/z of the isolated precursor in the spectrum is within the "method match tolerance" of the m/z of the precursor in your Skyline document.
You can change the method match tolerance at "Settings > Transition Settings > Instrument".
The default value of the method match tolerance is 0.055, but if you changed it to a larger number such as 0.2, then you will find that PRM works as an acquisition method.
You would not want to use "DIA/All Ions" for this data. That setting would only be appropriate for things like GC MS where the mass spectrometer fragmented everything without first filtering by precursor m/z.
If you hover the mouse over the Acquisition Method dropdown at "Settings > Transition Settings > Full Scan", Skyline will display a tooltip which tries to explain the difference between PRM, DIA, etc.
-- Nick |
| |
 Screenshot 2023-03-08 134016.png
Screenshot 2023-03-08 134016.png